生物信息学的工具箱
分析和可视化的基因和蛋白质图谱
Die生物信息学工具箱™bietet算法和Anwendungen für下一代测序(NGS),微阵列分析,massenspektrotrie和gen - ontology。mithilife von Toolbox-Funktionen können Sie genome and proteomische Daten ausstandarddateiformaten wie SAM, FASTA, CEL and CDF sowie aus在线- datenbanken wie dem NCBI基因表达Omnibus und GenBank®革命。Sie können diese Daten mit Sequenzbrowsern, räumlichen Heatmaps und Clusterprogrammen untersuchen und visualisieren。Die Toolbox bietet auch statistical Techniken zur Erkennung von Spitzenwerten, zur Unterstellung von Werten für fehlende Daten und zur Auswahl von Merkmalen。
Sie können Toolbox-Funktionen zur Unterstützung gängiger生物信息学Arbeitsabläufe kombinieren。Sie können ChIP-Seq-Daten verwenden, um transkriptionsaktoren zu identifizieren;RNA-Seq-Daten analysieren, um differentiell exprimierte Gene zu identifiieren;微阵列数据识别系统中变异的Kopienzahl和snp和蛋白质图谱麻省理工学院Hilfe von massenspektrometric - daten klassifizieren。
Erfahren Sie mehr überBioinformatik.
现在beginnen:
Die生物信息学工具箱enthält Algorithmen und Visualisierungstechniken für Die sequenzanalyze der nächsten Generation。模具工具箱ermöglicht es Ihnen,甘泽基因组祖分析人,während Sie Berechnungen auf einer Basispaar-Auflösungsebene durchführen。Sie können den NGS-Browser zur Visualisierung und Untersuchung von Short-Read-Ausrichtungen verwenden, wobei Sie entweder Single-End- oder pair - end - short Reads verwenden können。Sie können auch benutzerdefinierte Analyseroutinen erstellen, wie in den folgenden Beispielen gezeigt wild。
短读对齐可视化和Untersuchung des
mithilife des NGS-Browsers können Sie die Ausrichtung von Short-Read-Sequenzen überprüfen und untersuchen, um Analysen zur Messung der genetischen Variation and Genexpression zu unterstützen。Mit dem NGS-Browser können Sie:
- 短读daten一个einer Nukleotid-Referenzsequenz ausgerichtet visualisieren
- mehere Datensätze vergleichen, die an einer gemeinsamen Referenzsequenz ausgerichtet sinind
- 世界基础和地区民意调查结果
- Die Qualität und andere详细信息ausgerichteter Lesevorgänge untersuchen
- Diskrepanzen aufgrund von Basisaufruffehlern oder多形人鉴定
- Einfügungen und Löschungen可视化
- Merkmalsanmerkungen relativ zu einer bestimmten区域referenzsequenenabrufen

NGS-Browser, der Einzel-Nukleotid-Polymorphismen (SNPs) fettgedruckt anzeigt。Sie können mehere datenspren anzeigen, Spitzenwerte untersuchen, Einfügungen和Löschungen身份识别和死亡Lesequalität überprüfen。
Speichern和Verwalten von Short-Read-Sequenzdaten
Die bei der Sequenzanalyse der nächsten Generation verwendeten Datensätze sind für bestehende Speicherkapazitäten häufig zu umfangreich。Die Bioinformatics Toolbox bietet spezialisierte Datencontainer, mit denen Sie ganze Genome analyeren können。
麻省理工学院的民主党BioIndexedFile-Objekt können Sie auf den Inhalt von Textdateien zugreifen, die niht einheitlich groe Einträge wie Sequenzen, Anmerkungen und Querverweise auf den Datensatz enthalten。Sie können diese dijekte austabellen, flachen Dateien oder and wendungsspezifischen Formaten wie SAM, FASTA and FASTQ erzeeugen。
死BioMap-Klasse speichert Informationen aus Short-Read-Sequenzen, einschie ßlich Sequenz-Header, Lesesequenzen, Qualitätsbewertungen und Daten über Ausrichtung und Mapping auf eine einzige Referenzsequenz。Sie können物体和方法,在einem生物地图-物体和enthaltenen Daten zu untersuchen, auf Sie zuzugreifen, Sie zu filter和zu operpuliren。
微阵列
Sie können mehere Methoden zur Normalisierung von Microarray-Daten verwenden, einschließlich Lowess, globaler Mittelwert, mediane absolute Abweichung (MAD) und Quantilnormalisierung。Sie können diese Methoden auf den gesamten Microarray-Chip order auf bestimte region order Blöcke anwenden。麻省理工学院过滤和视觉分析技术können Sie Rohdaten vor der Ausführung von分析和视觉分析技术。
数据分析和可视化
Mit der生物信息学工具箱können Sie Hintergrundanpassungen vornehmen und die Expressionswerte von Genen (Sondensatz) aus Affymetrix®Microarray-Daten auf Sondenebene mit den Verfahren稳健多阵列平均(RMA)和GC稳健多阵列平均(GCRMA) berechnen。Sie können zirkuläre Binärsegmentierung auf Array-CGH-Daten anwenden und die falschfindunsrate von mehrfach假设bei der Prüfung von Genexpressionsdaten aus einem Microarray-Experiment schätzen。Sie können auch - eine秩不变集-正态化连接器für Sondenintensitäten für mehere Affymetrix ser - dateien order für Genexpressionswerte auszwei verschiedenen Versuchsbedingungen durchführen。
Microarrays - daten umfassen Vulkan-Plots, Box-Plots, log- plot, I-R-Plots und räumliche Wärmekarten des Microarrays。Sie können och表意文字mit g- band - musstervisualisieren。
mithillife von Routinen ausder统计和机器学习工具箱™können Sie Ihre Ergebnisse klassifizieren, hierarchische和K-Mittel-Clustering durchführen和Ihre Microarray-Daten in statischen Visualisierungen darstellen, z. B. 2D-Clustergramme mit optimaler Blattanordnung,热图,主成分图和Klassifizierungsbäume。

Vulkan-Plot von Microarray-Daten, die Signifikanz vs Genexpressionsverhältnis zeigen。
Die生物信息学工具箱bietet eine Reihe von Funktionen für Die分析von massenspektrometric - daten。Diese Funktionen ermöglichen die Vorverarbeitung, Klassifizierung和markkeridentifiierung aus SELDI-, MALDI-, LC/MS- und GC/MS- daten。Zu den Vorverarbeitungsfunktionen gehören Basislinienkorrektur, Glättung, Kalibrierung und Resampling。网址können德国激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光激光成像光谱仪durchführen。mehere Spektren können gleichzeeitig dargestellt werden。
Sie können Spektren glätten, ausrichten und normalisieren und dann Klassifikations und statisticsche lenwerkzeuge verwenden, um Klassifikatoren zu erstellen und potenzielle生物标志物zu identifiieren。

Kennzeichnungsfreie差异蛋白质组和代谢组分析mit der生物信息学工具箱。
石墨烯理论与可视化
Mit der生物信息学工具箱können Sie die grundlegende Graphentheorie auf dünn besetzte Matrizen anwenden。Sie können图画与漫画之间的互动,图画与漫画之间的层次,图画与漫画与漫画。Sie können kürzeste Wege in Diagrammen bestimmen and anzeigen, in gerichteten Diagrammen auf Zyklen testen und Isomorphie zwischen zwei Diagrammen finden。
统计与可视化
Die生物信息学工具箱bietet Funktionen, Die auf den klaassifizierungs and statischen Lernalgorithmen in der统计和机器学习工具箱Aufbauen, wie beispielsweise:
- 万博1manbetx支持向量机(SVM)和k-近邻- klassifikatoren
- 关于自由的实验和自由的信息,关于自由的klaassifizierungmethoden
- 交互工具zur Merkmalsauswahl,映射和Anzeige von层次图和Pfaden

统计与可视化。
Gen-Ontologie
Die生物信息学工具箱ermöglicht es Ihnen, aus MATLAB®在本体论-数据银行祖祖格里芬,本体论-kommentierte Dateien zu parsen和无本体论- wie Vorfahren, Nachkommen oder Verwandte zu erhalten。
Sequenzausrichtung
Die Toolbox bietet Funktionen, Objekte und Methoden für Die sequenzanalyze, einschließlich paarweiser Sequenz, sequenzproffil und Ausrichtung mehererer Sequenzen。大足华美gehoren:
- MATLAB - implementierungen von Standardalgorithmen für die lokale und global Sequenzausrichtung, wie z. B. Needleman-Wunsch-, Smith-Waterman- und profilversteckte markov - model - algorithmen
- 进步的Ausrichtung mehererer Sequenzen
- Grafische Darstellungen von Ausrichtungsergebnis-Matrizen
- 标准评分- matrixzen, wie z. B. die PAM- und BLOSUM-Matrixfamilien
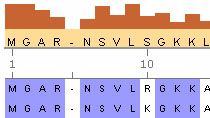
- Konsensus-Sequenzberechnung和Sequenzlogo-Anzeige

画廊(3 Bilder)
Dienstprogramme und statisticken für Sequenzen
Mit der Toolbox können Sie Ihre序列操作与分析器,um ein tieferes Verständnis Ihrer Daten zu erlangen。Sie können damit Folgendes tun:
- DNA-有序rna序列下的Verwendung des基因序列在Aminosäuresequenzen umwandeln
- Eine统计分析序列durchführen und nach bestimmten mussterinhalb einer Sequenz suchen。
- 重条带化酶和蛋白酶Durchführung von in- silicon - verdauung von Sequenzen oder zur Erstellung von Zufallssequenzen für Testfälle anwenden。
- Sekundärstruktur der RNA-Sequenzen mit minimaler freier Energie预后。
Sequenz-Visualisierung
模具工具箱ermöglicht es Ihnen,序列和Ausrichtungen zu可视化。Sie können lineare oder zirkuläre Karten von Sequenzen anzeigen, die mit genbank features versehen sind。Sie können Sekundärstrukturdiagramme einer RNA-Sequenz可视化。Mit interaktiven Betrachtungsgeräten können Sie paarweise und mehrfache Sequenzausrichtungen untersuchen and modifiziren。
Phylogenetische Baum-Analyse
Mit der Toolbox können Sie phylogenetische Bäume erstellen und bearbeiten。Sie können paarweise Abstände zwischen ausgerichteten oder nicht ausgerichteten Nukleotid- oder Aminosäuresequenzen under Verwendung einer breiten Palette von Ähnlichkeitsmetriken wie Jukes-Cantor, p-Abstand, align - wert oder einer benutzerdefinierten Abstandsmethode berechnen。系统遗传学Bäume werden unter Verwendung hierarchischer Verknüpfungen mit einer Vielzahl von Techniken konstruiert, darunter Nachbarschaftsverknüpfungen, Einzel und vollständige Verknüpfungen und UPGMA(未加权对组方法平均)。
模具工具箱unterstützt Die geichtung und das Umsetzen von Bäumen, Die Berechnung von Teilbäumen und Die Berechnung der kanonischen Form von Bäumen。系统发生学的起源können新事物的起源kürzen,新事物和新事物的起源,新事物和新事物的起源。Sie können auch die Anmerkungs-Tools in MATLAB verwenden,嗯Bäume in Präsentationsqualität zu erstellen。
分析蛋白蛋白
Die Toolbox bietet Techniken zur protein sequenzanalysis, einschließlich Routinen zur Berechnung von Eigenschaften einer Peptidsequenz wie atomare Zusammensetzung, isoelektrischer Punkt und molekulargeicht。Sie können die Aminosäurezusammensetzung von Proteinsequenzen bestimmen, ein Protein mit einem enzyme spalten und Backbone-Plots and Ramachandran-Plots von PDB-Daten erstellen。Sie können das序列工具verwenden, um die Eigenschaften einer Aminosäuresequenz anzuzeigen, oder den分子查看verwenden, um 3D-Molekularstrukturen anzuzeigen und zu manipulieren。
Dateiformate和Datenbankzugriff
Sie können auf Standarddateiformate für biologische Daten, Online-Datenbanken und Websites zugreifen。Mit der生物信息学工具箱können Sie:
- Sequenzdaten aus Standarddateiformaten, einschließlich FASTA, PDB und SCF lesen
- 微阵列daten aus Dateiformaten wie Affymetrix DAT-, EXP-, CEL-, CHP- und CDF-Dateien;Daten im ImaGene®-Ergebnisformat;安捷伦科技公司®特征提取软件- dateien;和GenePix®GPR和GAL-Dateien lesen
- Daten aus Online-Datenbanken wie GenBank, EMBL, NCBI BLAST和PDB lesen
- NCBI基因表达综合网站
- Zytogenetische Bandeninformationen us NCBI-Ideogrammen oder ucsc - zytoand - textdateien lesen
- massenspektrometer - daten ausmzxml和JCAMP-DX-Dateien lesen
Gemeinsame Nutzung von Algorithmen and Bereitstellung von Anwendungen
MATLAB bietet工具,mit denen Sie Ihr数据分析程序在eine maßgeschneiderte Softwareanwendung verwandeln können。大足gehören entwicklunstools zur Erstellung von Benutzeroberflächen, eine visuelle integrerte entwicklunsumgebung und ein Profiler。MATLAB-Produkte zur Anwendungsbereitstellung ermöglichen Ihnen die Integration Ihrer MATLAB-Algorithmen in bestehende Anwendungen in C, c++ and Java™,die Bereitstellung der entwickelten Algorithmen and benutzerdefinierten Schnittstellen als eigenständige Anwendungen, die Konvertierung von MATLAB-Algorithmen in Microsoft®.NET- oder COM-Komponenten, auf die von jder COM-basierten Anwendung aus zugegriffen werden kann, sowie die Erstellung von Microsoft Excel®-Add-Ins。
Sie können MATLAB在gängige Bioinformatik-Tools wie BioPerl, SOAP-basierte Webdienste和COM-Plugins integreren。

Gemeinsame Nutzung von Algorithmen and Bereitstellung von Anwendungen