癌症检测
这个例子展示了如何训练神经网络来检测癌症,使用蛋白质谱的质谱数据。
简介
血清蛋白质组学模式诊断可用于区分患有和没有疾病的患者的样本。使用表面增强激光解吸和电离(SELDI)蛋白质谱生成剖面模式。这项技术有可能改善癌症病理的临床诊断测试。
问题:癌症检测
目标是建立一个分类器,可以从质谱数据中区分癌症患者和对照组患者。
本例中采用的方法是选择一组减少的测量值或“特征”,使用分类器可用于区分癌症患者和对照组患者。这些特征是在特定质量/电荷值下的离子强度水平。
数据格式化
本例中使用的数据,在文件中提供ovarian_dataset.mat,来自FDA-NCI临床蛋白质组学项目数据库。该数据集的详细描述请参见[1]和[2]。
创建数据文件OvarianCancerQAQCdataset.mat按照下面的步骤利用序贯并行计算的光谱批处理(生物信息学工具箱).新文件包含变量Y,MZ,grp.
中的每一列Y表示取自患者的测量值。有216列Y代表216病人121卵巢癌患者和95都是正常病人。
每一行Y中所示的特定质量电荷值的离子强度水平MZ.有15000中质量荷值MZ每一行Y表示患者在特定质量电荷值下的离子强度水平。
的变量grp保存关于这些样本中哪些代表癌症患者,哪些代表正常患者的索引信息。
主要特性
该任务是一个典型的分类问题,其中特征的数量远远大于观测的数量,但单个特征实现了正确的分类。因此,我们的目标是找到一个分类器,它可以适当地学习如何对多个特征进行加权,同时产生一个不会过度拟合的广义映射。
寻找重要特征的一个简单方法是假设每个M/Z值是独立的,并计算一个双向t检验。rankfeatures返回最显著的M/Z值的索引,例如按检验统计量的绝对值排序的100个索引。
加载OvarianCancerQAQCdataset.mat并使用rankfeatures(生物信息学工具箱)选择100个排名最高的测量值作为输入x.
ind = rankfeatures(Y,grp,“标准”,的tt,“NumberOfIndices”, 100);x = Y(ind,:);
定义目标t这两个类别如下:
T = double(strcmp(“癌症”grp));T = [T;1 - t];
上面列出的脚本和示例中的预处理步骤旨在演示一组有代表性的可能的预处理和特征选择过程。使用不同的步骤或参数可能会导致不同的,可能更好的结果。
[x,t] = ovarian_dataset;谁xt
名称大小字节类属性t 2x216 3456 double x 100x216 172800 double
中的每一列x代表216个不同病人中的一个。
每一行x表示每个患者的100个特定质量电荷值中的一个的离子强度水平。
的变量t有两行216个值,每个值都是[1;0](表示癌症患者)或[0;1](表示正常患者)。
基于前馈神经网络的分类方法
现在您已经确定了一些重要的特征,您可以使用这些信息对癌症和正常样本进行分类。
由于神经网络是用随机的初始权值初始化的,所以每次运行示例时,训练网络后的结果都会略有不同。为了避免这种随机性,随机种子被设置为每次都重现相同的结果。但是,对于您自己的应用程序,设置随机种子并不是必需的。
setdemorandstream (672880951)
建立并训练具有5个隐层神经元的1隐层前馈神经网络。输入和目标样本自动分为训练集、验证集和测试集。训练集用于对网络进行教学。只要网络在验证集上不断改进,训练就会继续进行。测试集提供了网络精度的独立度量。
输入和输出的大小为0,因为网络还没有配置为匹配输入和目标数据。这种配置是在训练网络时进行的。
Net = patternnet(5);视图(净)
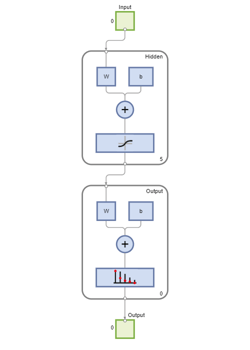
现在可以训练网络了。样本自动分为训练集、验证集和测试集。训练集用于对网络进行教学。只要网络在验证集上不断改进,训练就会继续进行。测试集提供了网络准确性的独立度量。
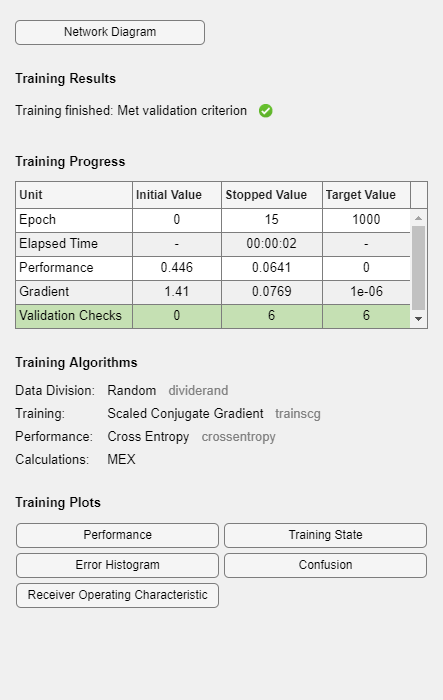
神经网络训练工具显示了正在训练的网络和用于训练它的算法。它还显示训练期间的训练状态,停止训练的标准以绿色突出显示。
底部的按钮可以打开有用的图,可以在训练期间和训练结束后打开。算法名称和绘图按钮旁边的链接打开了关于这些主题的文档。
[net,tr] = train(net,x,t);
要查看网络的性能在训练期间如何改进,请单击训练工具中的“性能”按钮,或使用plotperform函数。
性能以均方误差来衡量,并在对数刻度上显示。随着网络的训练,它迅速下降。
显示了每个训练集、验证集和测试集的性能。
plotperform (tr)

训练过的神经网络现在可以用我们从主数据集中划分的测试样本进行测试。测试数据没有以任何方式用于训练,因此提供了一个“样本外”数据集来测试网络。这给出了网络在使用来自现实世界的数据进行测试时的性能评估。
网络输出范围为0-1。将输出设置为1和0,分别表示癌症或正常患者。
testX = x(:,tr.testInd);testT = t(:,tr.testInd);testY = net(testX);testClasses = testY > 0.5
testClasses =2×32逻辑阵列1 1 1 1 1 1 1 1 1 1 1 1 1 0 0 1 1 0 0 1 1 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 0 1 1 0 0 1 1 1 1 1 1 1 1 1 0 1
衡量神经网络对数据拟合程度的一种方法是混淆图。
混淆矩阵显示了正确和不正确分类的百分比。正确的分类是矩阵对角线上的绿色方块。红色方块代表不正确的分类。
如果网络是准确的,那么红色方块中的百分比很小,表明很少有错误分类。
如果网络不准确,那么你可以尝试训练更长的时间,或者训练一个有更多隐藏神经元的网络。
plotconfusion (testT暴躁的)

这里是正确和错误分类的总百分比。
[c,cm] =困惑(testT,testY);流(正确分类:%f%%\n, 100 * (1 - c));
分类正确率:84.375000%
流('错误分类百分比:%f%%\n', 100 * c);
分类错误百分比:15.625000%
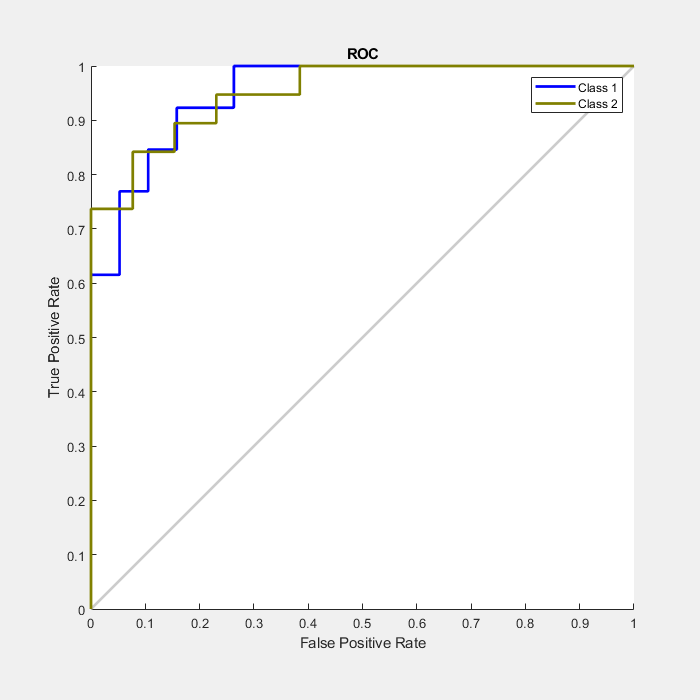
另一个衡量神经网络拟合数据的方法是接收者操作特征图。该图显示了当输出的阈值从0到1变化时,假阳性和真阳性率之间的关系。
直线越左越上,为了获得高的真阳性率,需要接受的假阳性就越少。最好的分类器有一条线,从左下角到左上角,再到右上角,或者接近这个位置。
1级为癌症患者,2级为正常患者。
plotroc (testT暴躁的)
这个例子展示了神经网络如何被用作癌症检测的分类器。为了提高分类器的性能,您还可以尝试使用主成分分析等技术来降低用于神经网络训练的数据的维数。
参考文献
[1] T.P. Conrads等,“高分辨率血清蛋白组学特征用于卵巢检测”,内分泌相关癌症,2004,11,pp. 163-178。
[2] E.F. Petricoin等,“使用血清蛋白质组学模式识别卵巢癌”,柳叶刀,359(9306),2002,pp. 572-577。
您也可以从以下列表中选择一个网站: