利用hmm进行一个蛋白质家族的剖面分析
这个例子展示了如何使用HMM配置文件来描述蛋白质家族。剖面分析是生物信息学的重要工具。常用的成对比较方法在分析远亲序列时通常不够敏感和特异性。相比之下,隐马尔可夫模型(HMM)概要文件提供了一种更好的选择,可以将查询序列与序列族的统计描述联系起来。HMM配置文件使用特定于位置的评分系统来捕获关于这些序列的多个对齐中不同位置的保守程度的信息。HMM谱分析可用于多个序列比对、数据库搜索、序列组成分析和模式分割,以及通过预测开放阅读框预测蛋白质结构和定位基因。
访问PFAM数据库
从一个已经构建好的蛋白质家族HMM开始这个例子。从桑格研究所数据库检索著名的7倍跨膜受体模型。PFAM密钥编号为PF00002。还检索用于训练该模型的预对齐序列。更多关于PFAM数据库的信息可以在http://pfam.xfam.org/.
Hmm_7tm = gethmmprof(2);种子_seqs =生殖恶性肿瘤(2,“类型”,“种子”);
为方便起见,以前下载的序列包含在mat文件中。注意,公共存储库中的数据经常被管理和更新;因此,当您使用最新的数据集时,本例的结果可能略有不同。
负载(“gpcrfam.mat”,“hmm_7tm”,“seed_seqs”)
模型和对齐也可以在以后直接从文件中存储和解析pfamhmmread,fastaread而且multialignread功能。
控件显示前三个加载序列的名称和内容seqdisp命令。
Seqdisp (seed_seqs([1 2 3]),“行”, 70)
ans = 23x81 char array '>VIPR2_HUMAN/123-371 ' ' 1 YILVKAIYTL GYSVS。LMSL atgsiilclf . rklhctr。N yihlnlflsf ilraisvlvk . ddvlysss。“71年GTLHCPD ... .......... .......... .... QPSSW。. . V。GCKLSL vflqyciman ffwllvegly ' '141 lhtllva ... ... mlpp。Rr cflaylligw glptvcigaw taar ...... .........我手机……'211 ...... tgc。WDTN。DHSVP W……WVIRI PILISIIVNF VLFISIIRIL LQKLT….SPDVGGNDQ SQY‘281 ....... .......... .......... ....Krlaks tllliplfgv hymv .. favf pisi…s。S' '351 kyqilfelcl gsf ....QGL vv ' ' ' '> vipr_carau /100-348 ' ' 1 frsvkigyti ghsvs。Lisl ttaivilcms . rklhctr。N YIHMHLFVSF ILKAIAVFVK .DAVLYDVIQ ESDNCS‘71 .... .......... .......... ..... TASV……Gckavi vffqycimas ffwllvegly ' '141 lhallavs .. ... ffse。Rk yfwwyiligw ggptifimaw sfak ...... .........一个YFND……' '211 ...... vgc。 WDIIENSDLF W....WIIKT PILASILMNF ILFICIIRIL RQKIN..... .CPDIGRNES' '281 NQY....... .......... .......... ....SRLAKS TLLLIPLFGI NFII..FAFI PENI...K.T' '351 ELRLVFDLIL GSF....QGF VV ' ' ' '>VIPR1_RAT/140-386 ' ' 1 YNTVKTGYTI GYSLS.LASL LVAMAILSLF .RKLHCTR.N YIHMHLFMSF ILRATAVFIK .DMALFNSG.' ' 71 EIDHCS.... .......... .......... .....EASV. ....GCKAAV VFFQYCVMAN FFWLLVEGLY' '141 LYTLLAVS.. ...FFSE.RK YFWGYILIGW GVPSVFITIW TVVR...... .........I YFED......' '211 ......FGC. WDTI.INSSL W....WIIKA PILLSILVNF VLFICIIRIL VQKLR..... .PPDIGKNDS' '281 SPY....... .......... .......... ....SRLAKS TLLLIPLFGI HYVM..FAFF PDNF...K.A' '351 QVKMVFELVV GSF....QGF VV '
关于如何在MATLAB®结构中存储配置文件HMM信息的更多信息,请参见帮助hmmprofstruct.
HMM对齐
要测试概要文件HMM对齐工具,您可以将多个对齐序列重新对齐到HMM模型。首先擦除用于格式化下载的对齐序列的序列中的句点。这样做将从序列中删除对齐信息。
Seqs = strrep({seed_seqs。序列},“。”,”);names = {seed_seq . header};
现在将所有蛋白质与HMM配置文件对齐。
流(“对齐序列”)分数=零(数字(seqs),1);Aligned_seqs = cell(数字(seqs),1);为sn = 1:元素个数(seq)流(“。”)[分数(sn) aligned_seqs {sn}] = hmmprofalign (hmm_7tm, seq {sn});结束流(' \ n ')
调整序列 ................................
接下来,将结果发送到Web浏览器,以便更好地探索新的多重对齐。底部标有*的列表示模型何时处于“匹配”或“删除”状态。
hmmprofmerge (aligned_seqs、名称、成绩)
您还可以从命令窗口探索对齐方式;的hmmprofmerge带有一个输出参数的函数将对齐的序列放入char数组中。
STR = hmmprofmerge(aligned_seqs);str(1:10,接触的
ans = 10x80 char array ' yilvkaiytlgysvs . lmslatgsiilclf . rklhrc . nyihlnlflsfilraisvlvk . ddvlysssg - tlh ......“FRSVKIGYTIGHSVS.LISLTTAIVILCMS.RKLHCTR.NYIHMHLFVSFILKAIAVFVK.DAVLYDVIQESDN……“YNTVKTGYTIGYSLS.LASLLVAMAILSLF.RKLHCTR.NYIHMHLFMSFILRATAVFIK.DMALFNSG-EIDH……“FGAIKTGYTIGHSLS.LISLTAAMIILCIF.RKLHCTR.NYIHMHLFMSFIMRAIAVFIK.DIVLFESG-ESDH……“YLSVKALYTVGYSTS.LVTLTTAMVILCRF.RKLHCTR.NFIHMNLFVSFMLRAISVFIK.DWILYAEQD-SSH……“FSTVKIIYTTGHSIS.IVALCVAIAILVAL.RRLHCPR.NYIHTQLFATFILKASAVFLK.DAAIFQGDS-TDH……“LSTLKQLYTAGYATS.LISLITAVIIFTCF.RKFHCTR.NYIHINLFVSFILRATAVFIK.DAVLFSDET-QNH……“FDRLGMIYTVGYSVS.LASLTVAVLILAYF.RRLHCTR.NYIHMHLFLSFMLRAVSIFVK.DAVLYSGATLDEA……“FERLYVMYTVGYSIS.FGSLAVAILIIGYF.RRLHCTR.NYIHMHLFVSFMLRATSIFVK.DRVVHAHIGVKEL……“ALNLFYLTIIGHGLS.IASLLISLGIFFYF.KSLSCQR.ITLHKNLFFSFVCNSVVTIIH.LTAVANNQALVAT……”
用序列比较寻找相似性
与普通序列比较相比,具有描述该家族的HHM配置文件有几个优点。假设你有一个新的寡核苷酸,你想把它与7-跨膜受体家族联系起来。在本例中,从NCBI中获取蛋白质序列并提取氨基酸序列。
Mousegpcr = getgenpept(“NP_783573”);Bai3 = mousegpcr.Sequence;
mat文件中也提供了这个序列gpcrfam.mat.
负载(“gpcrfam.mat”,“mousegpcr”) Bai3 = mousegpcr.Sequence;seqdisp (Bai3“行”, 70)
ans = 22x82字符数组' 1 MKAVRNLLIY IFSTYLLVMF GFNAAQDFWC STLVKGVIYG SYSVSEMFPK NFTNCTWTLE nppethsiy ' ' 71 LKFSKKDLSC SNFSLLAYQF DHFSHEKIKD LLRKNHSIMQ LCSSKNAFVF LQYDKNFIQI RRVFPTDFPG' ' 141 LQKKVEEDQK sffeflvspsqsqfgchk lspsqsqlesclk SENGRTESCG IMYTKCTCPQ HLGEWGIDDQ' ' ' 211 SLVLLNNVVL PLNEQTEGCL TQELQTTQVC NLTREAKRPP KEEFGMMGDH TIKSQRPRSV HEKRVPQEQA' ' ' 281 DAAKFMAQTG esgvewsqw SACSVTCGQG SQVRTRTCVS PYGTHCSGPL RESRVCNNTA lcpvvwee ' ' 351 WSPWSLCSFT CGRGQRTRTR SCTPPQYGGR PCEGPETHHK PCNIALCPVDGQWQEWSSWS HCSVTCSNGT' ' 421 QQRSRQCTAA AHGGSECRGP WAESRECYNP ECTANGQWNQ WGHWSGCSKS CDGGWERRMR TCQGAAVTGQ' ' 491 QCEGTGEEVR RCSEQRCPAP YEICPEDYLI SMVWKRTPAG DLAFNQCPLN ATGTTSRRCS LSLHGVASWE' ' 561 QPSFARCISN EYRHLQHSIK EHLAKGQRML AGDGMSQVTK TLLDLTQRKN FYAGDLLVSV EILRNVTDTF' ' 631 KRASYIPASD GVQNFFQIVS NLLDEENKEK WEDAQQIYPG SIELMQVIED FIHIVGMGMM DFQNSYLMTG' ' 701 NVVASIQKLP AASVLTDINF PMKGRKGMVD WARNSEDRVV IPKSIFTPVS SKELDESSVF VLGAVLYKNL' ' 771 DLILPTLRNY TVVNSKVIVV TIRPEPKTTD SFLEIELAHL ANGTLNPYCV LWDDSKSNES LGTWSTQGCK' ' 841 TVLTDASHTK CLCDRLSTFA ILAQQPREIV MESSGTPSVT LIVGSGLSCL ALITLAVVYA ALWRYIRSER' ' 911 SIILINFCLS IISSNILILV GQTQTHNKSI CTTTTAFLHF FFLASFCWVL TEAWQSYMAV TGKIRTRLIR' ' 981 KRFLCLGWGL PALVVATSVG FTRTKGYGTD HYCWLSLEGG LLYAFVGPAA AVVLVNMVIG ILVFNKLVSR' '1051 DGILDKKLKH RAGQMSEPHS GLTLKCAKCG VVSTTALSAT TASNAMASLW SSCVVLPLLA LTWMSAVLAM' '1121 TDKRSILFQI LFAVFDSLQG FVIVMVHCIL RREVQDAFRC RLRNCQDPIN ADSSSSFPNG HAQIMTDFEK' '1191 DVDIACRSVL HKDIGPCRAA TITGTLSRIS LNDDEEEKGT NPEGLSYSTL PGNVISKVII QQPTGLHMPM' '1261 SMNELSNPCL KKENTELRRT VYLCTDDNLR GADMDIVHPQ ERMMESDYIV MPRSSVSTQP SMKEESKMNI' '1331 GMETLPHERL LHYKVNPEFN MNPPVMDQFN MNLDQHLAPQ EHMQNLPFEP RTAVKNFMAS ELDDNVGLSR' '1401 SETGSTISMS SLERRKSRYS DLDFEKVMHT RKRHMELFQE LNQKFQTLDR FRDIPNTSSM ENPAPNKNPW' '1471 DTFKPPSEYQ HYTTINVLDT EAKDTLELRP AEWEKCLNLP LDVQEGDFQT EV '
首先,使用局部对齐将新序列与多个对齐中的一个序列进行比较。例如,使用第一个序列,在这种情况下,人类蛋白质“VIPR2”。Smith-Waterman算法(swalign)可以利用评分矩阵。打分矩阵可以捕捉符号替换的概率。我们知道这个例子中的序列只是远亲,所以BLOSUM30是评分矩阵的一个很好的选择。
VIPR2 = seqs{1};[sc_aa_affine, align] = swalign(Bai3,VIPR2,“ScoringMatrix”,...“blosum30”,“gapopen”5,“extendgap”3,“showscore”,真正的);sc_aa_affine
Sc_aa_affine = 69.6000
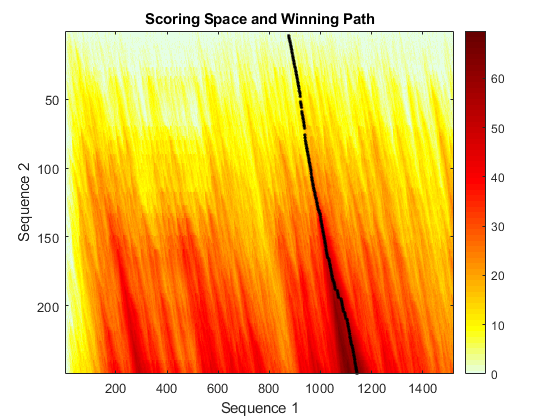
通过观察得分空间,显然这两个序列是相关的。然而,这种关系不能从点图中推断出来。
Bai3_aligned_region = strrep(align (1,:)),“- - -”,”);seqdotplot (VIPR2 Bai3_aligned_region 7, 2) ylabel (“VIPR2”);包含(“Bai3”);

这两个例子中的任何一个都足以证明这些序列是相关的吗?一种测试方法是随机创建一个氨基酸分布相同的假序列,看看它是如何与家族相匹配的。注意,假序列与VIPR2蛋白的局部比对得分并不明显低于Bia3与VIPR2蛋白的比对得分。为了确保这个例子结果的重现性,我们重置了全局随机生成器。
rng (0,“旋风”);fakeSeq = randseq(1000,“FROMSTRUCTURE”aacount (VIPR2));sc_fk_affine = swign (fakeSeq,VIPR2,“ScoringMatrix”,“blosum30”,...“gapopen”5,“extendgap”3,“showscore”,真正的)
Sc_fk_affine = 60.4000

相比之下,当您使用经过训练的配置文件HMM将两个序列与家族对齐时,将目标序列与家族配置文件对齐的得分明显大于将假序列对齐的得分。
sc_aa_hmm = hmmprofalign(hmm_7tm,Bai3) sc_fk_hmm = hmmprofalign(hmm_7tm,fakeSeq)
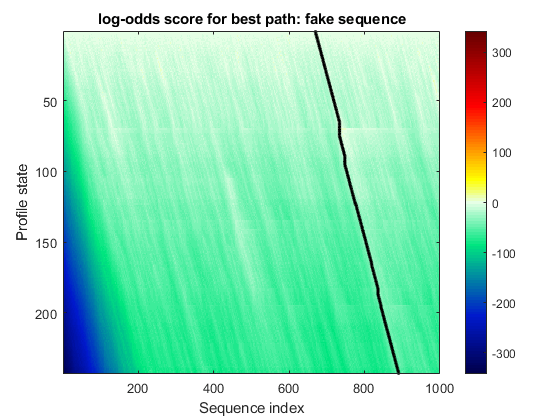
Sc_aa_hmm = 214.5286 sc_fk_hmm = -49.1624
探索概要HMM对齐选项
类似于swalign对齐功能,当您使用配置文件对齐时,您可以使用showscore选项。hmmprofalign函数。
显示Bai3对齐到7tm_2家族。
hmmprofalign (hmm_7tm Bai3,“showscore”,真正的);标题(最佳路径的对数赔率:Bai3);

显示与7tm_2家族对齐的“假”序列。
hmmprofalign (hmm_7tm fakeSeq,“showscore”,真正的);标题(“最佳路径的对数赔率分数:假序列”);
显示Bai3全局对齐到7tm_2家族。
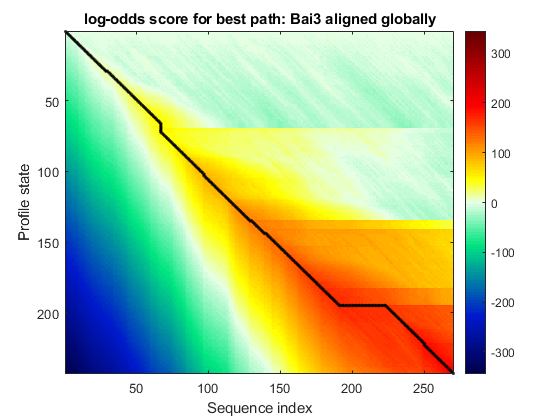
[sc_aa_hmm,align,ptrs] = hmmprofalign(hmm_7tm,Bai3);Bai3_hmmaligned_region = Bai3(min(ptrs):max(ptrs));hmmprofalign (hmm_7tm Bai3_hmmaligned_region,“showscore”,真正的);标题(“最佳路径的对数赔率得分:Bai3在全球范围内对齐”);
排列串联重复域。
naa = nummel (Bai3_hmmaligned_region);重复次数= randseq(1000,“FROMSTRUCTURE”aacount (Bai3));%人工例子repeats(200+(1:naa)) = Bai3_hmmaligned_region;repeats(500+(1:naa)) = Bai3_hmmaligned_region;repeats(700+(1:naa)) = Bai3_hmmaligned_region;hmmprofalign (hmm_7tm,重复,“showscore”,真正的);标题(“最佳路径的对数赔率分数:Bai3串联重复”);

搜索片段域
在MATLAB®中,您可以通过手动激活B - M >而且M - > EHMM模型的转换概率。
Hmm_7tm_f = hmm_7tm;hmm_7tm_f.BeginX(3:结束)= .002;hmm_7tm_f.MatchX (1: end-1, 4) = .002;
通过插入一小段Bai3蛋白,创建一个随机序列或片段模型:
片段= randseq(1000,“FROMSTRUCTURE”aacount (Bai3));fragment(501:550) = Bai3_hmmaligned_region(101:150);
尝试将随机序列与插入的肽对齐到全局模型和片段模型:
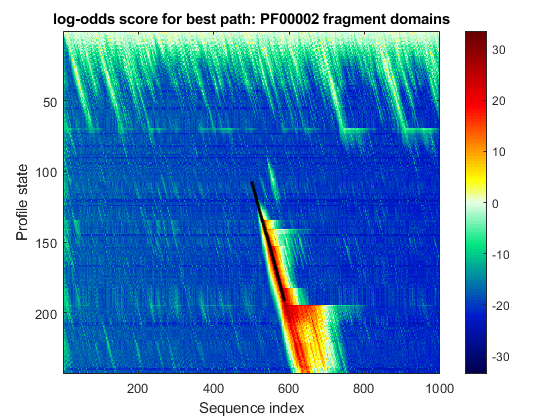
hmmprofalign (hmm_7tm片段,“showscore”,真正的);标题(“最佳路径的对数赔率得分:PF00002 global”);hmmprofalign (hmm_7tm_f片段,“showscore”,真正的);标题(“最佳路径的对数赔率得分:PF00002片段域”);

探索档案嗯
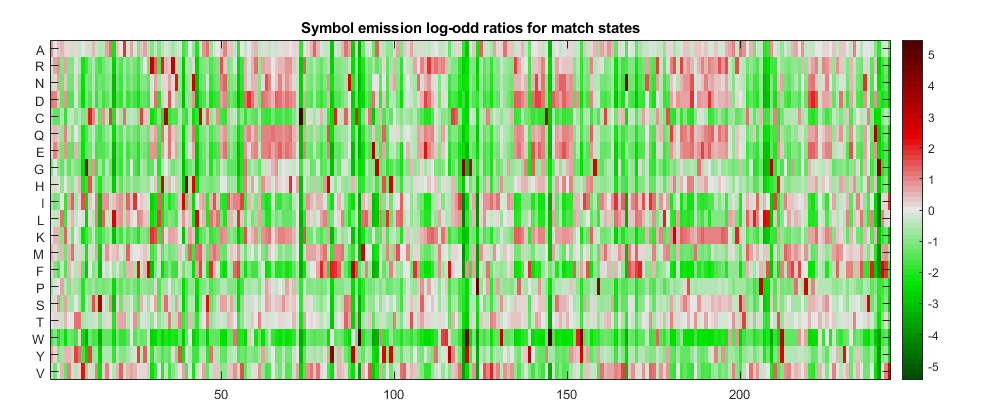
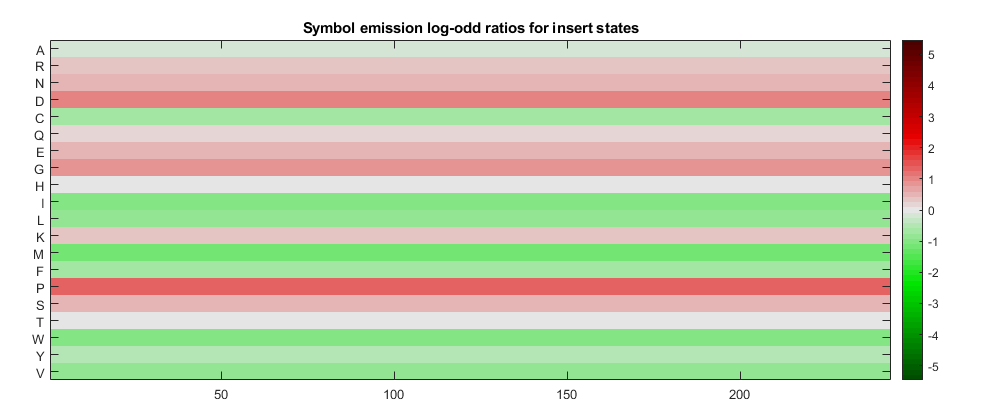
这个函数showhmmprof是一个探索概要文件HMM的交互式工具。尝试用鼠标左右点击模型图形。每个模型有三个图:(1)符号发射概率匹配状态,(2)符号发射概率在插入州,和(3)过渡概率。
showhmmprof (hmm_7tm“规模”,“logodds”)

探索概要文件HMM的另一种方法是从多个对齐中创建一个序列徽标。序列标志显示给定区域内每个位置发现的碱基的频率,通常用于绑定位点。使用hmm_7tm序列,考虑在PTRR_Human序列的n端发现的甲状旁腺激素相关肽受体(前体)的部分。的seqlogo可以快速直观地比较该区域在7tm家族中的保存情况。
seqlogo (str,“startat”,1,“endat”, 20岁,“字母”,“AA”)

概要文件的评估
轮廓hmm也可以通过多次对准来估计。随着与族相关的新序列的发现,有可能重新估计模型参数。
Hmm_7tm_new = hmmprofestimate(hmm_7tm,str)
hmm_7tm_new = struct with fields: Name: '7tm_2' PfamAccessionNumber: 'PF00002.19' ModelDescription: '7跨膜受体(分泌素家族)' ModelLength: 243字母表:'AA' matchemmission: [243x20 double] insertemmission: [243x20 double] NullEmission:[0.0768 0.0418 0.0396 0.0305 0.0201 0.0378…] BeginX: [244x1 double] MatchX: [242x4 double] InsertX: [242x2 double] DeleteX: [242x2 double] FlankingInsertX: [2x2 double] LoopX: [2x2 double] NullX: [2x1 double]
如果您的序列没有预先对齐,您还可以使用multialign函数,然后再估计新的HMM概要文件。可以通过将序列重新对齐到模型并迭代地重新估计模型来改进HMM概要,直到收敛到局部最优模型。
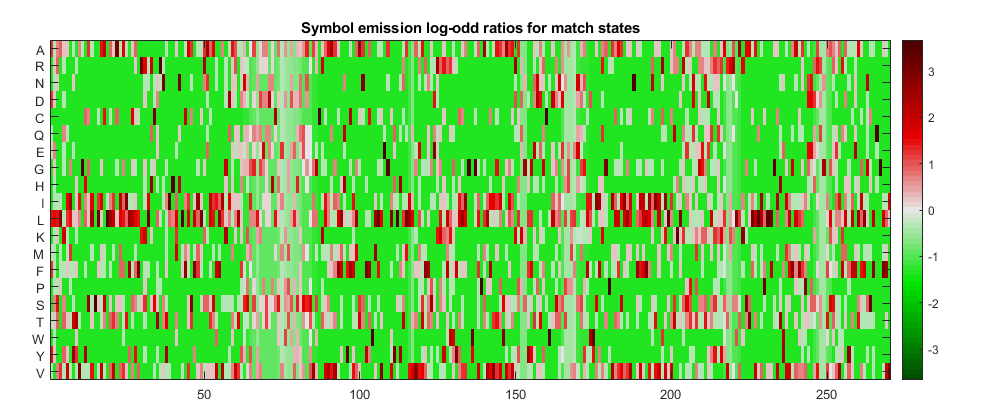
Aligned_seqs = multialign(seqs);Hmm_7tm_ma = hmmprofestimate(hmmprofstruct(270), alignd_seqs) showhmmprof(Hmm_7tm_ma,“规模”,“logodds”)关闭;关闭;%关闭插入发射问题。还有过渡问题。
hmm_7tm_ma = struct with fields: ModelLength: 270 Alphabet: 'AA' matchemmission: [270x20 double] insertemmission: [270x20 double] NullEmission:[0.0500 0.0500 0.0500 0.0500 0.0500 0.0500 0.0500 0.0500 0.0500 0.0500…] BeginX: [271x1 double] MatchX: [269x4 double] InsertX: [269x2 double] DeleteX: [269x2 double] FlankingInsertX: [2x2 double] LoopX: [2x2 double] NullX: [2x1 double]
将所有序列对齐到新模型。
流(“对齐序列”)分数=零(数字(seqs),1);Aligned_seqs = cell(数字(seqs),1);为sn = 1:元素个数(seq)流(“。”)[分数(sn) aligned_seqs {sn}] = hmmprofalign (hmm_7tm_ma, seq {sn});结束流(' \ n ') STR = hmmprofmerge(aligned_seqs);str(1:10,接触的
调整序列 ................................ans = 10x80 char array 'YILVKAIYTLGYSVSLMSLATGSIILCLF.RKLHCTRNYIHLNLFLSFILRAISVLVKDDVLYSS——SGTLHCP-....“FRSVKIGYTIGHSVSLISLTTAIVILCMS.RKLHCTRNYIHMHLFVSFILKAIAVFVKDAVLYDVIQ——ESDNCS -…“YNTVKTGYTIGYSLSLASLLVAMAILSLF.RKLHCTRNYIHMHLFMSFILRATAVFIKDMALFNS——GEIDHCS -…“FGAIKTGYTIGHSLSLISLTAAMIILCIF.RKLHCTRNYIHMHLFMSFIMRAIAVFIKDIVLFES——GESDHCH -…“YLSVKALYTVGYSTSLVTLTTAMVILCRF.RKLHCTRNFIHMNLFVSFMLRAISVFIKDWILYAE——QDSSHCF -…“FSTVKIIYTTGHSISIVALCVAIAILVAL.RRLHCPRNYIHTQLFATFILKASAVFLKDAAIFQG——DSTDHCS -…“LSTLKQLYTAGYATSLISLITAVIIFTCF.RKFHCTRNYIHINLFVSFILRATAVFIKDAVLFSD——ETQNHCL -…“FDRLGMIYTVGYSVSLASLTVAVLILAYF.RRLHCTRNYIHMHLFLSFMLRAVSIFVKDAVLYSGATLDEAERLTE……“FERLYVMYTVGYSISFGSLAVAILIIGYF.RRLHCTRNYIHMHLFVSFMLRATSIFVKDRVVHAHIGVKELESLIM……“ALNLFYLTIIGHGLSIASLLISLGIFFYF.KSLSCQRITLHKNLFFSFVCNSVVTIIHLTAVANNQALVATNP——……”
在帮助浏览器中显示对齐的序列。
hmmprofmerge (aligned_seqs、名称、成绩)
您也可以从以下列表中选择一个网站: