Bioinformatics工具箱
对基因组和蛋白质组进行可视化分析
Bioinformatics Toolbox™Contiene Algoritmi E APP每IL Shequenziamento di Nuova Generazione(NGS,下一代测序),L'Analisi di MicroArray,La Spettrometria di Massa E L'Ontologia Genica。Grazie Alle Funzioni del Toolbox,è可能leggere dati genomici exceomici da formati文件标准sam,fasta,cel e cdf,oltre che da data database在线来到Il Gene表达omnibus dell'ncbi e Genbank®. È在序列图中,在calore spaziali e clustergram的地图中,可能会出现可视化问题。在皮奇市的技术统计局,瓦洛里市的自然资源中心是一个特色城市。
Positibile Combinare Le Funzioni del Toolbox Per S万博1manbetxhiteSare Flussi di Lavoro di Bioinformaticapiùdiffusi。èCosibibleUSARE Dati Chip-SEQ每个人I Fattori di Trascrizione;Analizzare I Dati RNA-SEQ每个人I Geni Interprisemente浓咖啡;Trovare Le Varianti Del Numero di Copie E在Dati di MicroArray中的Gli SNP;Classificare I Profili Delle Proteine usando dati di spettrometria di massa。
马吉奥利·因莫西奥尼·苏拉计算机生物学。
乌拉岛:
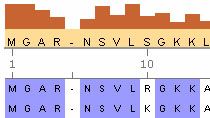
生物信息学工具箱提供新一代生物分析的算法和技术(NGS)。我的工具箱一致同意进行基因组间的分析。È可能的usare il browser NGS per visualizzare e studiare gli allineamenti delle read corte usando read corte di tipo single-end or paired-end。È anche可能的服装套路分析个性化,来说明忽略esempi che guono。
visual zione e studio dell 'allineamento delle reads corte
我们可以通过浏览NGS, è来验证我们的研究,我们可以对基因序列进行分析,我们可以对基因序列进行分析。万博1manbetxIl浏览器NGS同意di:
- Visualizzare dati di读取Corte Allineati A Una Sequenza di Riferimento Nucleotidica
- 在所有冲突中,对抗都是以不同的方式进行的
- 想象不同的基础区域和不同的序列
- 研究大蒜的质量
- 个人在政治基础上的错误分类
- 视觉化eliminazioni
- Recuperare annotazioni迪功能相对于UNA大区的specifica德拉SEQUENZA迪riferimento

浏览器NGS,在Cui Vengono Illustrati dei Polimorfismi在Gransetto中的单晶核苷酸(SNP)。èChissibileVisualizzare不同的Tracciati di Dati,esaminare我picchi,个人格雷尼·伊斯特利斯e le eliminazioni e studiasrelaqualitàdelle读书。
ConservazioneèGESTIONE代DATI阿尔sequenze二读科尔特
我根据《金融记忆》的《新一代金融分析》设定了数据效用。生物信息学工具箱提供了一份关于基因内部分析的同意书。
L'oggetto生物indexedfile.同意在测试中加入所有内容或文件,且内容和尺寸不一致的情况下,按照数据集的顺序、注释和检验。任务Oggeti possono essere生成一份表格,平面文件,格式特定,根据确定的应用程序。SAM,FASTA e FASTQ。
内拉·克拉斯BioMap我们可以将所有的信息顺序读给corte,将所有的信息顺序读给corte,将所有的信息顺序读给qualità,将所有的信息顺序读给qualità,将所有的信息顺序读给真主安拉,将所有的信息顺序读给真主安拉,将所有的信息顺序读给È可能的效用proprietà di oggetti e metodi per esplorare, accedere,滤镜e manipolare i dati contenuti in un oggetto BioMap。
Normalizzazione di微阵列
èConsibibileUSARE Diversi Metodi Per Qualizzare I Dati Di Microarray,ComplosaLacomenizzazione Lowess,Della Media Globale,Della Deviazione Mediana Assoluta(Mad)e La Normalizzazione Stime。Questi Metodi Possone Essere Applati All'Intero Chip Del MicroArray O区域o Blocchi特定。Le Funzioni di Filtraggio E Stationuzione Consenterono di ulire i dati grezzi prima di eseSuire le Analisi e Le rustine divestiveizzione。
数据可视化分析
生物信息工具箱consente二eseguire regolazioni德尔背景E双链calcolare我valori二espressione DEI GENI(集合二探空仪)一个partire岱DATI一个livello二雄达二微阵列的Affymetrix®鲁棒多阵列平均(RMA)和GC鲁棒多阵列平均(GCRMA)。È可能的应用是一个节段的圆形的数据阵列- cgh e stimare il tasso di falsa scoperta di più test di ipotesi dei dati di意式数据阵列和一个部分的精子微阵列。在più, è可能eseguire la normalizzazione a rango invariante sulle intensità delle sonde per più文件CEL Affymetrix o sui valori di espressione de geni a partire due condizioni sperimenti diverse。
Le常规专业专长每拉宫桂节dei dati dei microarray comand comono i火山图,i broadloplot,i grafici log-log,i grafici i-r e le mappe di Calore spaziali del microarray。èChePossibileVisualizzareDilegli Idegography Con Pattern Di G-anding。
我的例行公事统计和机器学习工具箱™例如,可能的分类法、聚类法、K-均值法、可视化统计中的基因芯片数据表示法、聚类图2D比对法、calore地图、主成分分析法和分类法。

基因表达谱中的火山图。
生物信息工具箱offre联合国一套双funzioni每L'analisi代DATI迪spettrometria迪马萨。Queste funzioni consentono二预elaborare,classificareÈidentificare我marcatori哒DATI SELDI,MALDI,LC / MSëGC / MS。勒funzioni二预elaborazione comprendono LA correzione,啦linearizzazione,LA calibrazioneÈIL ricampionamento DEI DATI二基。Èanche不可能性allineare我DATI德利阿布鲁spettri grezzi山岛L'ASSE M / Zëprocedere all'allineamento德尔节奏二ritenzioneスDATI LC / MSëGC / MS。È不可能性eseguire IL plottaggio迪彪spettri contemporaneamente。
GLI spettri possono essere linearizzati,allineatiēnormalizzati。POI,感恩教堂的Agli工具类迪classificazioneēapprendimento statistico,SI possono creare代classificatoriËindividuare代potenziali biomarcatori。

analisi metabolomica e proteomica不同endiale senza Etichette Con Bioinformatics工具箱。
Teoria代grafiËvisualizzazione
生物信息学工具箱同意将grafi基础应用于矩阵稀疏。È可能的creare,可视化的grafi和manipolare grafi come le mappe di interazione,i grafici gerarchi i pathway。在确定是否符合格拉菲标准的视觉化和渗透性的仪器上,验证是否符合格拉菲标准。
Apprendimimeno Statistico E Visualizzione
生物信息工具箱offre funzioni车operano sugli algoritmi迪classificazioneËapprendimento statistico迪统计和机器学习工具箱,崔茶:
- Classificatori支万博1manbetx持向量机(SVM)e K-Interlight邻居
- 每个组织都进行了交叉验证,以确定是否有不同的分类
- 根据选择功能的跨部门仪器,地图和视觉化grafici Gerarchi路径

视觉化统计。
Ontologia杰妮卡
生物信息学工具箱permette di Concere al数据库del progetto Gene Ontology da MATLAB®,di analizzare i文件安宁蒂迪纳洛洛世Genica e di Ottienere dei sottogruppi dell'ontologia来到Gli Antenati,我Discendenti o我hentri。
Allineamento di sequenze
我的工具箱里有一个funzioni的配置,可以对序列进行分析,对一个副本进行综合的序列,对序列进行多重的分析。园子温compresi:
- 实施MATLAB di算法标准,以满足全球各地的需求,包括Needleman Wunsch的l'algoritmo、Smith Waterman e quello del modello nascosto di Markov
- 对多个进行性序列进行排序
- 我画了一幅画,画了一个轮廓线
- 标准基质,来自于基质PAM e BLOSUM家族
- Calcolo Delle Sequenze Consenso E Visualizzione del Logo Della Shequenza

埃斯普洛拉广场(伊马基尼3号)
Statisticheè效用相对全部sequenze
我的工具箱同意分析每个理解的正确序列。E的可行性:
- Convertire sequenze二DNAöRNA在sequenze二amminoacidi山岛IL codice genetico
- 对特定的尾蚴模式进行统计分析和序列分析
- 根据casi di试验,在硅片序列或硅片序列中使用酶限制和蛋白质
- prevedere la struttura secondaria con il minimo dell'energia libera delle sendenze di Rna
Visualizzazione delle sequenze
工具库同意对所有序列进行可视化。工具库可能对序列进行线性或环形标注。工具库可能对RNA序列进行二次可视化。工具库可能对所有序列进行可视化昆兹:我要一个铜币。
analisi di Alberi丝瘤
我的工具箱一致认为我是阿尔贝丝状体。È可能的calcolare le distance tra le coppie, tra sequize di nucleotidi o aminoacidi allineate o meno, usando un ' amia gamma di metriche di iglianza, come Jukes-Cantor, p-distance, alignment-score o altro metodo definito dall 'utente。Unweighted Pair Group Method, UPGMA (Unweighted Pair Group Method, Unweighted Pair Group Method, Unweighted Pair Group Method, UPGMA)。
工具箱支持计算比索和阿万博1manbetx尔贝里半径,计算子树和规范形式。在波塔雷同意的丝状纤维形成术的可视化仪器中,有一台是riordinare e rinominare i rami;esplorare le distanze;leggere o scrivere文件格式为di Newick。根据目前的情况,在MATLAB中显示gli仪器的可能性。
analisi delle carateristiche delle proteine
IL Toolbox Offre Tecniche Per Analizzare Le Sendenze di Proteine,Compres Perle常规Per IL CalcoloProprietàdiUna Sequenza di Peptidi来La Composizione Dell'atomo,Il Punto Isoelettrico E IL PESO Sollarare。è可能确定洛杉矶Composizione Degli Amminoacidi Delle Sequenze Di Proteine,Scindere Una Proteina Con Un Enzima E Craree Graficii Del骨架E Grafici di Ramachandran di Dati Pdb。è可能usare inl序列工具per vedere leproprietàdi una sequenza di Amminoacidi o Il分子观众每个Visualizzare e Manipolare Strutture Sollatulari 3D。
文件访问数据库的格式
È possible accedere a formati file standard per I dati biologici, a database online e a siti web。生物信息学工具箱同意di:
- Leggere我DATI阿尔sequenze达formati文件标准来FASTA,PDBēSCF
- Leggere I Dati Dei MicroArray D Formati文件来我文件Affymetrix DAT,EXP,CEL,CHP E CDF;Dati di Risultati Imagene®;文件安捷伦®特征提取软件;文件GenePix®探地雷达
- Leggere I Dati da数据库在线来到Genbank,Embl,NCBI Blast E PDB
- 基因表达的重要数据综合性数据
- Leggere客房信息BANDE citogenetiche dagli ideogrammi dell'NCBIØ大appositi文件迪德图dell'UCSC
- 马萨诸塞州斯佩特罗梅里亚的Leggere dai文件MZXML e JCAMP-DX
条件是分配应用的算法
MATLAB提供了在应用软件个性化中对数据进行程序分析的工具。在使用界面的时候,我们不能使用压缩的乐器,也不能使用集成的乐器和分析器。I prodotti per la distribuzione delle applicazioni MATLAB consentono di integrare I propri algoritmi MATLAB concodice C, c++ e applicazioni Java™esistenti, di distribuire gli algoritmi svilupati e interface personalizate come applicazioni standalone, di convertire gli algoritmi MATLAB in componenti Microsoft®.Net o com a cui sipuòachaceraqualsiasi应用程序苏基com e di crahe加载项di microsoft Excel®。
è可以整容Matlab Con Gli Strumenti di BioinformaticaPiùUSATI来生物培养器,I Servizi Web Basati Su Soap E I插入COM。

Condivisione di Algoritmi E Distribuzione Di Applicazioni。