生物信息学工具箱
读取、分析和可视化基因组和蛋白质组数据
生物信息工具箱™提供算法和应用程序用于下一代测序(NGS),微阵列分析,质谱法,和基因本体论。使用工具箱功能,可以读取标准的文件格式,如SAM,FASTA,CEL和民防部队,以及来自在线数据库如NCBI基因表达综合和GenBank基因组和蛋白质组数据®。您可以探索和可视化与序列浏览器,空间热图,并clustergrams这个数据。该工具箱还提供了用于检测的峰,对于缺失数据插补值,以及选择特征的统计技术。
您可以组合工具箱功能来支持常见的生物信息学工作流。万博1manbetx你可以使用ChIP-Seq数据来识别转录因子;分析RNA-Seq数据,识别差异表达基因;识别芯片数据中的拷贝数变体和snp;并利用质谱数据对蛋白质谱进行分类。
了解更多关于计算生物学。
开始:
生物信息学工具箱提供了用于下一代测序分析的算法和可视化技术。工具箱使您能够分析整个基因组,同时以碱基对的分辨率执行计算。您可以使用NGS浏览器使用单端或双端短读来可视化和研究短读对齐。您还可以构建自定义分析例程,如下面的示例所示。
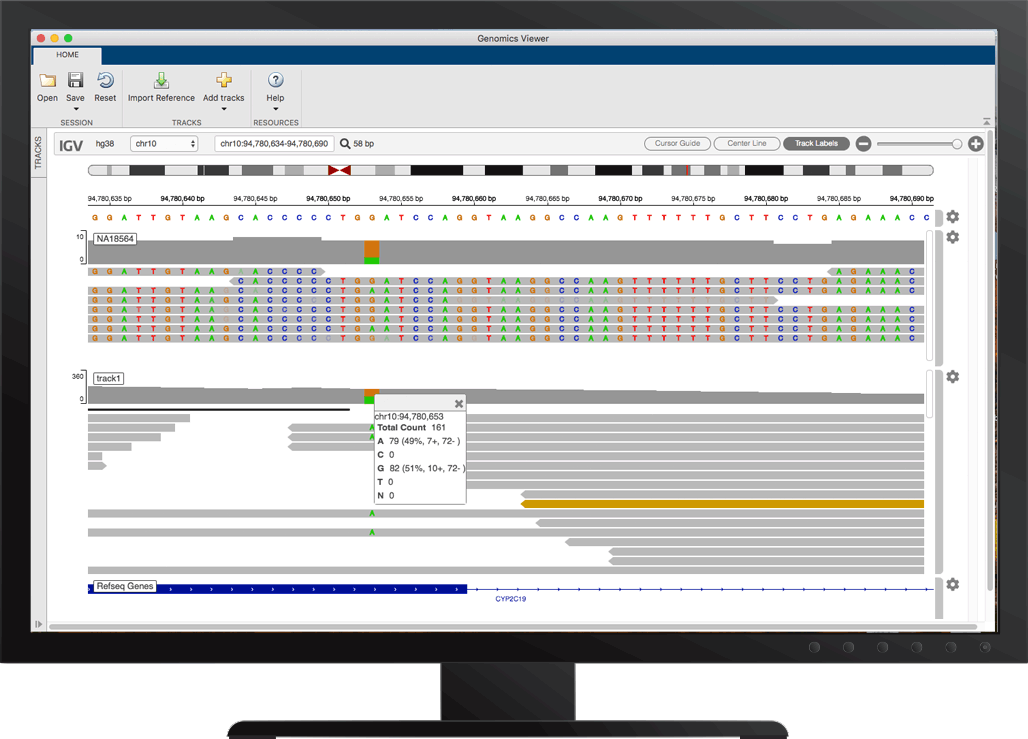
可视化和调查短读对齐
使用NGS浏览器,您可以验证和调查短读序列的排列,以支持测量遗传变异和基因表达的分析。万博1manbetxNGS浏览器让你:
- 可视化与核苷酸参考序列对齐的短读数据
- 比较按公共引用序列对齐的多个数据集
- 查看参考序列的不同基底和区域的覆盖范围
- 调查质量和对准读取其他细节
- 识别由于基础调用错误或多态性造成的不匹配
- 可视化的插入和缺失
- 检索与引用序列的特定区域相关的特征注释

NGS浏览器,以粗体显示单核苷酸多态性(SNPs)。您可以显示数据的多个轨迹,检查峰值,识别插入和删除,并检查读取质量。
存储和管理短读序列数据
在下一代测序分析中使用的数据集通常太大,无法成物理存储器。生物信息学工具箱提供专业的数据容器,使您能够分析整个基因组。
该BioIndexedFile对象允许您访问文本文件的内容,其中包含对数据集的不一致大小的条目,如序列、注释和交叉引用。您可以从表、平面文件或特定于应用程序的格式(如SAM、FASTA和FASTQ)生成这些对象。
该BioMap类存储来自短读序列的信息,包括序列标头、读序列、质量分数,以及关于对齐和映射到单个参考序列的数据。您可以使用对象属性和方法来探索、访问、筛选和操作BioMap对象中包含的数据。
芯片正常化
可以使用几种方法对微阵列数据进行归一化,包括低值、全局均值、中位数绝对偏差(MAD)和分位数归一化。您可以将这些方法应用于整个微阵列芯片或特定的区域或块。过滤和输入函数允许您在运行分析和可视化例程之前清理原始数据。
数据分析和可视化
生物信息学工具箱让您执行背景调整和计算基因(探针集)表达值从Affymetrix®微阵列探针级数据使用稳健多阵列平均(RMA)和GC稳健多阵列平均(GCRMA)程序。可以将循环二值分割应用于阵列CGH数据,并通过一个微阵列实验对基因表达数据进行多重假设检验,估计其误发现率。您还可以对多个Affymetrix CEL文件或来自两个不同实验条件的基因表达值的探测强度对秩不变集进行归一化。
可视化微阵列数据的专用例程包括火山图、盒图、loglog图、I-R图和微阵列的空间热图。您还可以用g带模式可视化表意文字。
从使用例程统计和机器学习工具箱™,您可以对结果进行分类,执行分层和K-means聚类,并以统计可视化的方式表示微阵列数据,例如具有最佳叶序的2D聚类图、热图、主成分图和分类树。

微阵列数据火山图显示基因表达率与基因表达率的关系。
生物信息学工具箱提供了一套用于质谱数据分析的功能。这些功能支持对SELDI、MALDI、LC/MS和GC/MS数据进行预处理、分类和标识。预处理功能包括基线校正、平滑、校准和重采样。您可以使用M/Z轴对齐原始光谱数据,并在LC/MS和GC/MS数据上执行保持时间对齐。你可以同时绘制多个光谱。
您可以平滑、对齐和标准化光谱,然后使用分类和统计学习工具来创建分类器和识别潜在的生物标记

使用生物信息学工具箱进行无标记的差异蛋白质组学和代谢组学分析。
图论与可视化
生物信息工具箱使您能够基本图形理论应用到稀疏矩阵。您可以创建,查看和操作的图形,如相互作用图谱,层次结构图和途径。您可以确定并查看有向图的循环图中的最短路径,测试和两个图形之间找到同构。
统计学习与可视化
生物信息学工具箱提供的功能是建立在分类和统计学习算法统计和机器学习工具箱,包括:
- 万博1manbetx支持向量机(SVM)和K近邻分类器
- 设立交叉验证实验和测量不同的分类方法的性能功能
- 用于特征选择、映射和显示层次图和路径的交互式工具

统计学习和可视化。
基因本体论
生物信息学工具箱使您能够从MATLAB中访问基因本体数据库®,解析基因本体注解文件,并且获得这样的本体的子集作为祖先,后代,或亲戚。
序列比对
工具箱提供用于序列分析的函数、对象和方法,包括成对序列、序列概要和多重序列比对。这些包括:
- 用于局部和全局序列比对的标准算法的MATLAB实现,如Needleman-Wunsch、Smith-Waterman和profile-hidden Markov模型算法
- 渐进多序列对准
- 对齐结果矩阵的图形表示
- 标准得分矩阵,如PAM和BLOSUM矩阵家庭
- 共有序列计算与序列标识显示

探索画廊(3个图像)
序列实用程序和统计数据
该工具箱让你能够操作和分析您的序列,以获得您的数据更深入的了解。您可以:
- 利用遗传密码将DNA或RNA序列转换为氨基酸序列
- 对序列进行统计分析,并在序列中搜索特定的模式
- 应用限制性内切酶和蛋白酶对序列进行有机硅消化或为测试用例创建随机序列
- 预测RNA序列的最小自由能二级结构
序列可视化
工具箱使您能够可视化序列和对齐。您可以查看带有GenBank特性注释的序列的线性或圆形映射。你可以看到RNA序列的二级结构图。交互式查看器允许您探索和修改成对和多个序列对齐。
系统进化树分析
该工具箱使您能够创建和编辑系统进化树。就可以计算出对齐或不对齐的核苷酸或使用宽范围的相似性度量,例如鸠克斯-坎托,对距离,对齐得分,或用户定义的距离方法的氨基酸序列之间的成对距离。系统发育树使用了各种技术层次链接,包括邻居加入,单一的,完整的联动构造,和不加权对组平均法(UPGMA)。
该工具箱支撑加权和rer万博1manbetxooting树木,计算子树,并计算树木的规范形式。进化树浏览器可以让你修剪,重新排序和重命名分支机构;探索距离;和读取或写入Newick格式的文件。您也可以使用注释工具在MATLAB中创建演示文稿质量树。
蛋白质功能分析
该工具箱提供蛋白质序列分析技术,包括例程,用于计算诸如原子组成,等电点和分子量的肽序列的性质。可以测定蛋白质序列的氨基酸组合物,裂解蛋白的酶,并创建骨干图和PDB数据的拉氏图。可以使用序列工具来查看的氨基酸序列的性质或使用分子查看器来显示和操纵3D分子结构。
文件格式和数据库访问
您可以访问生物数据、在线数据库和Web站点的标准文件格式。生物信息学工具箱使您能够:
- 从标准文件格式中读取序列数据,包括FASTA、PDB和SCF
- 从文件中读取格式,如Affymetrix公司DAT,EXP,CEL,热电联产,以及CDF文件芯片的数据;ImaGene®结果格式数据;安捷伦科技公司®特征提取软件的文件;和的GenePix®GPR和GAL文件
- 从GenBank、EMBL、NCBI BLAST和PDB等在线数据库读取数据
- 使用单个命令直接从NCBI基因表达综合网站导入数据
- 从NCBI表意文字或UCSC细胞带文本文件中读取细胞遗传条带信息
- 从MZXML和JCAMP-DX文件中读取质谱数据
共享算法和部署应用程序
MATLAB提供了一些工具,可以让您将数据分析程序转换为定制的软件应用程序。这些工具包括用于构建用户界面的开发工具、可视化集成开发环境和分析器。MATLAB应用程序部署产品允许您将MATLAB算法与现有的s manbetx 845C、c++和Java集成™应用程序,部署已开发的算法和自定义接口作为独立的应用程序,将MATLAB算法转换成Microsoft®可以从任何基于COM的应用程序访问的。net或COM组件,并创建Microsoft Excel®加载项。
您可以将MATLAB与常用的生物信息学工具(如BioPerl、基于soap的Web服务和COM插件)集成在一起。

共享算法和部署应用程序。
