识别显着的特征和分类蛋白质谱
此示例显示了如何对质谱数据进行分类并使用一些统计工具寻找潜在的疾病标记和蛋白质组学模式诊断。
介绍
血清蛋白质组学模式诊断可用于区分来自患者和不含疾病的患者。使用表面增强的激光解吸和电离(SELDI)蛋白质质谱法产生谱模式。该技术有可能改善癌症病理学的临床诊断测试。目标是选择可用于区分癌症和对照患者的减少的测量或“特征”集。这些特征将是特定质量/充电值的离子强度水平。
预处理数据
在此示例中设置的卵巢癌数据是来自FDA-NCI临床蛋白质组学计划数据库。使用WCX2蛋白质阵列产生数据集。数据集包括95个控制和121个卵巢癌。有关此数据集的详细说明,请参见[1]和[4]。
此示例假定您已拥有预处理数据ovararcancerqaqcdataset.mat。但是,如果您没有数据文件,则可以通过遵循示例中的步骤来重新创建使用顺序和并行计算的光谱批处理。
或者,您可以运行脚本Msseqprocessing.m.。
AddPath(FullFile(MatlaBroot,'例子'那'bioinfo'那'主要的')))%确保支持文件在搜索路径上万博1manbetx类型MSSEQPROCESSING.
%MSSeqProcessing脚本以创建ovararcancerqaqcdataset.mat(用于%CancerDetectDemo)。在运行此文件之前,将变量%“存储库”初始化为放置您放置质量频谱%文件的完整路径。例如:%% repository ='f:/ masspecropository / ovararcd_postqaqc /';%%或%repository ='/ home / username / masspecropository / ovararcd_postqaqc /';%%执行近似时间为18分钟(奔腾4,4GHz)。如果%%有并行计算工具箱,请参阅BioDistCompdemo以查看%如何加速此分析。%Copyright 2003-2008 MathWorks,Inc. RespositoryC = [存储库'癌症/'];repositoryn = [存储库'普通/'];filescancer = dir([repositoryc' * .txt']);numbercancerdatasets = numel(粉丝); fprintf('Found %d Cancer mass-spectrograms.\n',NumberCancerDatasets) filesNormal = dir([repositoryN '*.txt']); NumberNormalDatasets = numel(filesNormal); fprintf('Found %d Control mass-spectrograms.\n',NumberNormalDatasets) files = [ strcat('Cancer/',{filesCancer.name}) ... strcat('Normal/',{filesNormal.name})]; N = numel(files); % total number of files fprintf('Total %d mass-spectrograms to process...\n',N) [MZ,Y] = msbatchprocessing(repository,files); disp('Finished; normalizing and saving to OvarianCancerQAQCdataset.mat.') Y = msnorm(MZ,Y,'QUANTILE',0.5,'LIMITS',[3500 11000],'MAX',50); grp = [repmat({'Cancer'},size(filesCancer));... repmat({'Normal'},size(filesNormal))]; save OvarianCancerQAQCdataset.mat Y MZ grp
从脚本和上面列出的示例的预处理步骤旨在说明一组可能的预处理过程。使用不同的步骤或参数可能导致该示例的不同和可能改善的结果。
加载数据
一旦您拥有预处理的数据,就可以将其加载到MATLAB中。
加载ovararcancerqaqcdataset.谁是
名称大小字节类属性MZ 15000x1 120000双y 15000x216 25920000 Double Grup 216x1 25056细胞
有三个变量:MZ.那y那GRP.。MZ.是质量/充电矢量,y是所有216名患者(对照和癌症)的强度值,以及GRP.将索引信息持有以下哪些样本代表癌症患者,哪些代表正常患者。
初始化通过示例使用的一些变量。
n = numel(grp);%样本数量cidx = strcmp('癌症',GRP);癌症样本的%逻辑指数载体nidx = strcmp('普通的',GRP);普通样本的%逻辑索引矢量cvec =查找(cidx);癌症样本的%指数载体nvec = find(nidx);普通样本的%索引向量Xaxislabel =.'质量/充电(m / z)';%x的图标标签yaxislabel ='离子强度';%Y标签的图表
可视化一些样品
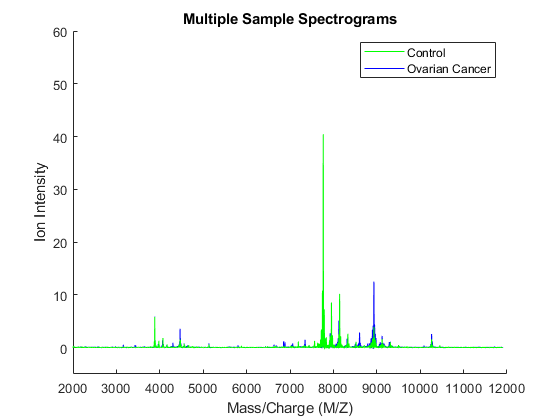
您可以将一些数据集绘制到图形窗口中,以便在视觉上比较两组的配置文件;在该实施例中,显示来自癌症患者(蓝色)和来自对照患者(绿色)的五种谱图。
数字;抓住在;HC = Plot(MZ,Y(:,CVEC(1:5)),'B');hn = plot(mz,y(:,nvec(1:5)),'G');Xlabel(Xaxislabel);ylabel(yaxislabel);轴([2000 12000 -5 60])图例([HN(1),HC(1)],{'控制'那'卵巢癌'}) 标题('多个样本谱图')
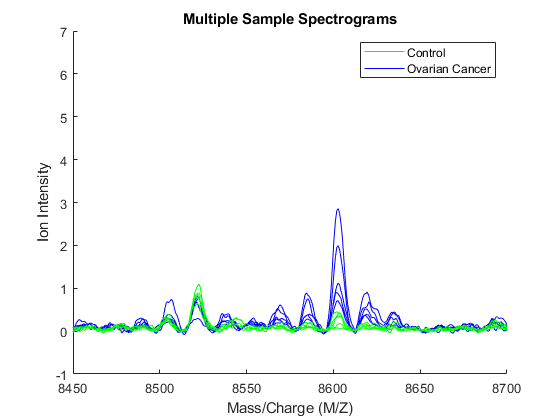
放大到8500至8700 M / z的区域上显示了一些可用于分类数据的峰值。
轴([8450,8700,-1,7])
可视化整个数据集的另一种方法是查看控制和癌症样本的组平均信号。您可以绘制组平均值和每个组的信封。
mean_n =均值(y(:,nidx),2);对照样品的%组平均值max_n = max(y(:,nidx),[],2);对照样品的百分比包络min_n = min(y(:,nidx),[],2);对照样品的%底部包络mean_c =均值(y(:,cidx),2);癌症样本的%群体平均值max_c = max(y(:,cidx),[],2);对照样品的百分比包络min_c = min(y(:,cidx),[],2);对照样品的%底部包络数字;抓住在;hc = plot(mz,mean_c,'B');hn = plot(mz,mean_n,'G');gc = plot(mz,[max_c min_c],'B--');gn = plot(mz,[max_n min_n],'G - ');Xlabel(Xaxislabel);ylabel(yaxislabel);轴([8450,8700,-1,7])图例([HN,HC,GN(1),GC(1)],{'控制组AVG。'那'卵巢癌群AVG'那......'控制信封'那'卵巢癌信封'},......'地点'那'西北') 标题('平均水平和集团信封')

观察显然没有单一的功能可以完美地歧视两个群体。
排名主要特征
用于查找重要特征的简单方法是假设每个M / Z值都是独立的并且计算双向T检验。randfeatures.将索引返回到最高有效的m / z值,例如由测试统计的绝对值排名的100个索引。此特征选择方法也称为过滤方法,其中学习算法不涉及如何选择特征。
[壮举,stat] = rantfeatures(y,grp,'标准'那'ttest'那'数字',100);
第一个输出randfeatures.可用于提取有效功能的M / Z值。
sig_masses = mz(feat);sig_masses(1:7)'%显示前七
ANS = 1.0E + 03 * 8.1009 8.1016 8.1024 8.1024 8.1001 8.1032 7.7366 7.7359
第二个输出randfeatures.是一个带有测试统计数据的绝对值的矢量。您可以使用光谱绘制它yyaxis.。
数字;yyaxis.剩下绘图(MZ,[MEAN_N MEAL_C]);ylim([ - 1,20])xlim([7950,8300])标题('重要的m / z值')Xlabel(Xaxislabel);ylabel(yaxislabel);yyaxis.对绘图(MZ,统计数据);ylim([ - 1,22])ylabel('测试统计');传奇({'控制组AVG。'那'卵巢癌群AVG。'那'测试统计'})
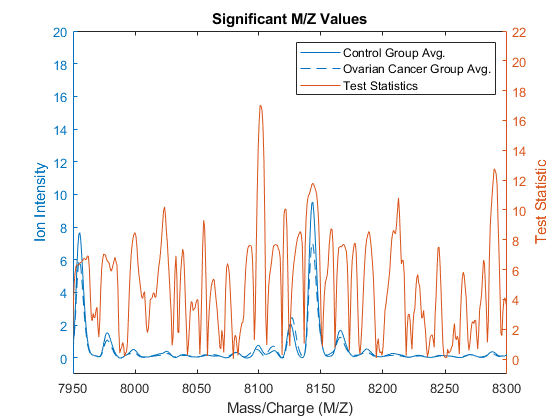
请注意,高m / z值下有很大的区域,但强度低(〜8100 da)。测量类别可分离性的其他方法可用randfeatures.,例如基于熵,Bhattacharyya,或经验接收器操作特征(Roc)曲线的区域。
使用线性判别分析(LDA)盲分类
既然您已识别出一些重要的功能,您可以使用此信息对癌症和正常样本进行分类。由于样本数量少,您可以使用20%HoldOut运行交叉验证,以更好地估计分类器性能。CVPartition.允许您为不同类型的系统评估方法设置培训和测试索引,例如保持,k折叠和留下。
per_eval = 0.20;交叉验证的%训练大小RNG('默认');%将随机生成器初始化到同一状态%用于生成已发布的示例cv = cvpartition(grp,'坚持',per_eval)
CV =保持交叉验证分区NUMOBSERATIONS:216 NumTestSets:1列塔:173 Testsize:43
观察该功能仅从训练子集中选择,并且使用测试子集执行验证。Classperf.允许您跟踪多种验证。
cp_lda1 = classperf(grp);%初始化CP对象为了k = 1:10%运行交叉验证10次cv = redartition(cv);Feat = RankFeatures(Y(:,培训(CV)),GRP(培训(CV)),'数字',100);c =分类(y(feat,test(cv))',y(feat,培训(cv))',grp(培训(cv)));ClassPerf(CP_LDA1,C,测试(CV));%使用当前验证更新CP对象结尾
循环后,您可以使用CP对象中的任何属性评估整体盲分类的性能,例如错误率,灵敏度,特异性等。
cp_lda1.
标签:''''''''''comparesslabels:{2x1 cell} toundtruth:[216x1 double] numberofobservations:216 ControlClasses:2 TargetClasses:1验证分布:10个采样分布:[216x1双]错误作用:[216x1双] SampleDistRibutionByClass:[2x1双]ErrorDistributionByClass:[2×1双] CountingMatrix:[3×2双] CorrectRate:0.8488 ERRORRATE:0.1512 LastCorrectRate:0.8837 LastErrorRate:0.1163 InconclusiveRate:0 ClassifiedRate:1灵敏度:0.8208特异性:0.8842 PositivePredictiveValue:0.8995 NegativePredictiveValue:0.7962 PositiveLikelihood:7.0890 NegativeLikelihood:0.2026患病率:0.5581诊断:[2x2双]
通过基于区域信息消除一些特征,可以提高这种朴素的特征选择方法。例如,'nweight'中randfeatures.超过相邻M / Z特征的测试统计,使得其他有效的M / Z值可以合并到所选功能的子集中
cp_lda2 = classperf(grp);%初始化CP对象为了k = 1:10%运行交叉验证10次cv = redartition(cv);Feat = RankFeatures(Y(:,培训(CV)),GRP(培训(CV)),'数字',100,'nweight'5);c =分类(y(feat,test(cv))',y(feat,培训(cv))',grp(培训(cv)));ClassPerf(CP_LDA2,C,测试(CV));%使用当前验证更新CP对象结尾cp_lda2.correctrate.%平均正确分类率
ans = 0.9023
PCA / LDA减少数据维度
Lilien等人。介绍[2]算法减少使用主成分分析(PCA)的数据维度,然后使用LDA来对组进行分类。在此示例中,M / Z空间中最重要的功能的2000中映射到150主组件
cp_pcalda = classperf(grp);%初始化CP对象为了k = 1:10%运行交叉验证10次cv = redartition(cv);%选择2000个最重要的功能。Feat = RankFeatures(Y(:,培训(CV)),GRP(培训(CV)),'数字',2000);%PCA减少维度p = pca(y(feat,培训(cv))');%项目进入PCA空间x = y(feat,:)'* p(:,1:150);%使用LDA.c =分类(x(测试(cv),:),x(培训(cv),:),grp(培训(cv)));classperf(cp_pcalda,c,test(cv));结尾cp_pcalda.correctrate.%平均正确分类率
ANS = 0.9814.
随机搜索子集特征选择
特征选择也可以通过分类加强,这种方法通常被称为包装选择方法。随机搜索特征选择生成随机功能,并与学习算法独立评估其质量。稍后,它选择一个最常见的良好功能的池。李等人。在[3]中,将该概念应用于蛋白质表达模式的分析。这兰特菲特功能允许您使用LDA或K最近邻分类在随机的特征子集中搜索特征的子集。
注意:以下示例是计算密集的,因此它已从示例中禁用。此外,为更好的结果,您应该从默认值增加池大小和分类器的严格性兰特菲特。类型帮助Randfeatures.了解更多信息。
如果0.%<==更改为1以启用。这可能会使用广泛的时间来完成。cv = redartition(cv);[Feat,Fcount] = RandFeatures(Y(y:,培训(cv)),grp(培训(cv)),......'分类器'那'da'那'performancethreshold',0.90);别的加载RandFeatCancerDetect.结尾
使用评估集评估所选功能的质量
第一个输出来自兰特菲特是一个有序的MZ值指数列表。第一项在达到良好分类的子集中最常出现。第二个输出是每个值选择的次数的实际计数。您可以使用hist看这个分布。
数字;hist(fcount,max(fcount)+1);
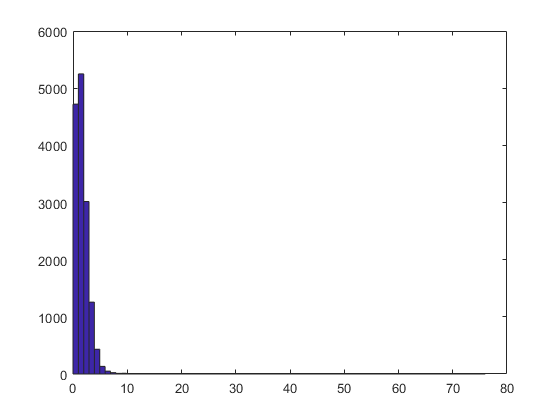
您将看到大多数值最多在选定的子集中出现。放大更好地了解更频繁选定值的详细信息。
轴([0 80 0 100])

只有几个值选择超过10次。您可以通过使用step图来显示这些来显示最常见的选定功能。
数字;抓住在;sigfeats = fcount;sigfeats(sigfeats <= 10)= 0;绘图(MZ,[MEAN_N MEAL_C]);茎(MZ(Sigfeats> 0),Sigfeats(Sigfeats> 0),'r');轴([2000,12000,-1,80])传奇({'控制组AVG。'那'卵巢癌群AVG。'那'重要的功能'},......'地点'那'西北')Xlabel(Xaxislabel);ylabel(yaxislabel);
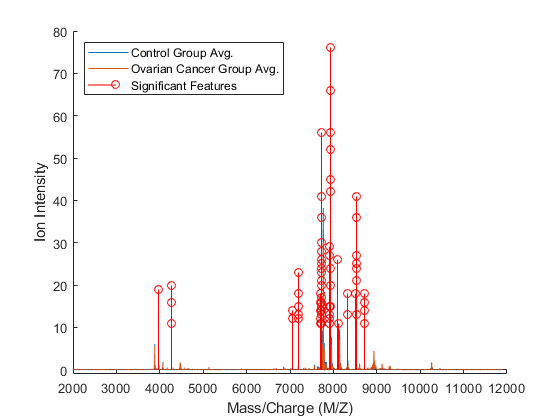
这些功能似乎在几个组中聚集在一起。您可以通过运行以下实验,进一步调查多少功能很重要。最常见的选择功能用于对数据进行分类,然后使用两个最常用的选定功能等,直到使用超过10次的所有功能。然后,您可以看看添加更多功能是否可提高分类器。
nsig = sum(fcount> 10);cp_rndfeat = zeros(20,nsig);为了我= 1:nsig为了j = 1:20 cv = redartition(cv);p = pca(y(feat(1:i),培训(cv))');x = y(feat(1:i),:)'* p;c =分类(x(测试(cv),:),x(培训(cv),:),grp(培训(cv)));cp = classperf(grp,c,test(cv));cp_rndfeat(j,i)= cp.correctate;%平均正确分类率结尾结尾图绘图(1:nsig,[max(cp_rndfeat);平均值(cp_rndfeat)]);传奇({“最好的钢筋”那'均值曲折'},'地点'那'东南')
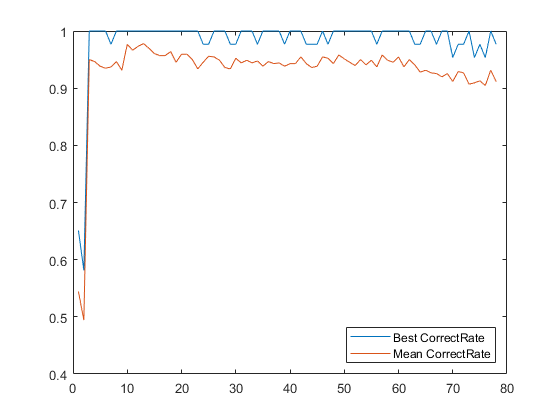
从这个图表中,您可以看到,只要有三个特征即可获得完美的分类。您还会注意到,对于少量功能,最大的正确率的最大值发生,然后逐渐减少。
[BestaVerageCR,Bestnumfeatures] = Max(平均值(cp_rndfeat));
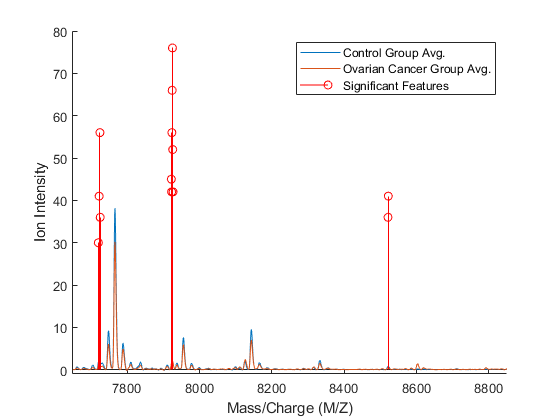
您现在可以可视化提供最佳平均分类的功能。您可以看到这些实际上对应于数据中的三个峰值。
数字;抓住在;sigfeats = fcount;sigfeats(sigfeats <= 10)= 0;ax_handle = plot(mz,[mean_n mean_c]);茎(MZ(壮举(1:Bestnumfeatures)),Sigfeats(Feat(1:Bestnumfeatures)),'r');轴([7650,8850,-1,80])图例({'控制组AVG。'那'卵巢癌群AVG。'那'重要的功能'})Xlabel(Xaxislabel);ylabel(yaxislabel);
替代统计学习算法
Matlab®中有许多分类工具,您也可以用于分析蛋白质组学数据。其中包括支持向量机(万博1manbetxfitcsvm.),K-最近的邻居(Fitcknn.),神经网络(深度学习工具箱™)和分类树(fitctree)。对于功能选择,您还可以使用顺序子集功能选择(序列)通过使用遗传算法(全局优化工具箱)来优化随机搜索方法。例如,看遗传算法在质谱数据中搜索特征。
参考
[1]Conrads,T P,V A Fusaro,S Ross,D Johann,V Rajapakse,B A Hitt,S M Steinberg等。“卵巢癌检测的”高分辨率血清蛋白质组学特征“。内分泌相关癌症,2004年6月,163-78。
[2]Lilien,Ryan H.,Hany Farid,以及Bruce R. Donald。“人血清质谱”依赖于表达蛋白质组学数据的概率疾病分类。“计算生物学10,NO。6(2003年12月):925-46。
[3]Li,L.,D. M.Mubach,P.Terry和J. A. Taylor。“GA / KNN方法在Seldi蛋白质组学数据中的应用。”生物信息学20,不。10(2004年7月1日):1638-40。
[4]百素,Emanuel F,Ali M Ardekani,Ben A Hitt,Peter J Levine,Vincent A Fusaro,Seth M Steinberg,Gordon B Mills等。“在血清中使用蛋白质组学模式来鉴定卵巢癌。”柳叶瓶359,没有。9306(2002年2月):572-77。
也可以看看
Classperf.|msnorm.|randfeatures.
相关话题
您还可以从以下列表中选择一个网站: