使用贝叶斯分析基于数组的全息数据隐马尔科夫模型
这个例子展示了如何使用一个贝叶斯隐马尔可夫模型(HMM)技术确定拷贝数改变基于数组的比较基因组杂交(CGH)数据。
介绍
基于数组的计算全息是一种高通量的技术来测量整个基因组DNA拷贝数变化。DNA片段或“克隆”的测试和参考样品杂化映射数组片段。Log2强度比率测试参考提供有用信息的全基因组资料拷贝数。在理想情况下,log2比正常(copy-neutral)克隆log2 (2/2) = 0单拷贝的损失log2 (1/2) = 1,单副本收益log2 (3/2) = 0.58。多个副本收益或放大的值log2 (4/2),log2 (5/2),....两个副本,或删除对应的值负。在真实的应用程序中,即使考虑了测量误差,log2比率差别很大从理论价值。通常比例缩小为零。一个主要因素是正常细胞的肿瘤样本的污染。还有一个依赖强度比率之间的邻近克隆。这些问题需要使用有效的统计算法描述基因组资料。
贝叶斯嗯
古et al .,(2006)提出了一种贝叶斯统计方法根据隐马尔科夫模型(HMM)用于分析全息阵列数据。隐马尔科夫模型占相邻克隆之间的依赖。强度比率是由隐藏的拷贝数。贝叶斯学习是用来识别全基因组拷贝数变化的数据。后的推论是拷贝数的收益与损失。
在这个贝叶斯HMM算法,有四个州,定义为拷贝数损失状态(1),拷贝数中性状态(2),单一副本获得状态(3)和放大(多个获得)状态(4)。与每个克隆拷贝数状态相关联。规范化log2比率假定分布

的μ是一个未知参数为每个国家这个约束:

先知先觉的拷贝数的变化是:



![$ $ [\ mu_4 | \ mu_3 \ sigma_3] \ sim N (1 \ tau_4 ^ 2) \ cdot我(\ mu_4& # 62; \ mu_3 + & # xA; 3 \ sigma_3) $ $](http://www.tianjin-qmedu.com/help/examples/bioinfo/win64/acghhmmdemo_eq02275008633568004220.png)
古et al .,(2006)还描述了一种Metropolis-within-Gibbs算法生成后样品。每个染色体的密度算法是独立运营的染色体产生密度样品的参数。的起始值参数产生的先知先觉。生成的拷贝数州代表从边际利益后,对于每个密度画,生成的状态检查和归类为焦点的替代品,过渡点,放大,异常值和全染色体的变化。
在本例中,您将贝叶斯HMM算法来分析一些胰腺癌的全息阵列配置文件样本[2]。
加载数据
这个示例中的数据是24的全息阵列配置文件胰腺癌细胞系和13原发肿瘤标本Alguirre et al ., (2004)。标记DNA片段杂交在安捷伦®包含14160 cDNA克隆人类的互补脱氧核糖核酸微阵列。大约9420个克隆有独特的地图位置映射元素之间的平均时间间隔为100.1 kb。可以找到更详细的数据和实验[2]。为了方便,数据已经规范化和log2基础强度比率由垫提供文件pancrea_oligocgh.mat。
您将应用贝叶斯HMM算法分析染色体12胰腺腺癌样本6的数据,并比较结果与部分发现的圆形二元分割(CBS)算法Olshen et al ., (2004)。
加载包含log2强度比率和垫文件映射37个基因位置的样本。
负载pancrea_oligocghpancrea_data
pancrea_data =结构体字段:示例:{37 x1细胞}染色体:[13446 x1 int8] GenomicPosition: [13446 x1 int32] Log2Ratio: [13446 x37双]Log2RatioMed: [13446 x37双]Log2RatioSeg: [13446 x37双]cloneid: [13446 x1 int32]
指定染色体数目和样本分析。
sampleIndex = 6;chromID = 12;示例= pancrea_data.Sample {sampleIndex}
示例= ' PA.C.Dan.G '
加载和绘制log2比12号染色体的数据样本PA.C.Dan.G。
idx = pancrea_data。染色体= = chromID;双(X = pancrea_data.GenomicPosition (idx));Y = pancrea_data。Log2Ratio (idx sampleIndex);%去除南数据点idx = ~ isnan (Y);X = X (idx);Y = Y (idx);%画出数据图;情节(X, Y,“。”,“颜色”,0.6 0.6 1)ylims = [-1.5, 3.5];ylims ylim (gca)标题(sprintf (' % s -染色体% d '、样本、chromID))包含(“基因组的位置”);ylabel (“Log2(比率)”)

12号染色体上的克隆数量进行分析
N =元素个数(Y)
N = 437
执行循环二元分割
你可以通过执行染色体分析分割使用CBS算法[3],这是实现cghcbs函数。这个过程需要花费数秒。您可以查看的情节部分指的是通过指定的原始数据SHOWPLOT参数。注意:你可以输入医生cghcbs对这个函数的更多细节。
PS = cghcbs (pancrea_data,“SampleInd”sampleIndex,…“染色体”chromID,“ShowPlot”,chromID);ylims ylim (gca)
分析:PA.C.Dan.G。目前12号染色体

如图,CBS过程声明的集合高强度比率作为两个独立的部分。CBS过程还发现一个地区与拷贝数的损失。
初始化参数
贝叶斯HMM方法使用Metropolis-within-Gibbs算法生成后的样品参数[1]。模型参数分为四个街区。该算法迭代生成的四个条件对剩下的块和数据块。
与贝叶斯HMM算法分析数据,您需要初始化参数。更多细节前参数可以在参考文献[1]和[4]。
初始化随机数发生器的状态,以确保这些命令生成的数据匹配数据的HTML版本,这个例子。
rng (“默认”);
定义的状态数
NS = 4;
定义密度的迭代的数量
NMC = 100;
确定hyperparameters先验分布的四个州。
mus_hyper = [1 0、0.58、1];taus_hyper = (1, 1, 1, 2);
设置参数ε决定的约束手段。
每股收益= 0.1;
设置边界前的每个状态的手段。
mu_low_bounds =(负无穷,每股收益,每股收益,0.58);mu_up_bounds =(每股收益、每股收益0.58,正);
古et al .,(2006)假定先验误差方差的倒数(σ^ 2)作为州伽马分布下界为0.41 1、2和3。设置规模参数γ分布为每个状态。
sg_alpha = [1 1 1 1];sg_beta = (1, 1, 1, 1);sg_bounds = (0.41 0.41 0.41 1);
定义一个变量州存储拷贝数状态序列的克隆获得每个迭代。
州= 0 (N, NMC);
定义一个变量st_counts来保存状态转换为每个拷贝数计数状态。
st_counts = 0 (NS, NS);
确定先验分布
获得迭代开始
iloop = 1;
确定的四个州(伽马分布的抽样与以前的规模参数α和β。
(= 0 (NS, NMC);为i = 1: NS(开关)(我,iloop) = acghhmmsample (“伽马”sg_alpha (i), sg_beta(我),sg_bounds (i));结束
确定方法,四个州的采样截断正态分布上下边界之间的意思。注:第四下界将取决于第三状态。
亩= 0 (NS, NMC);为我= 1:NS如果我= = 4 mu_low_bounds(4) =σ亩(3,iloop) + 3 * (3, iloop);结束亩(我,iloop) = acghhmmsample (“正常”mus_hyper (i), taus_hyper(我),…mu_low_bounds(我),mu_up_bounds (i));结束
承担独立的狄利克雷先验随机4 x4转移概率矩阵的行[1]。生成随机转移矩阵从之前狄利克雷分布。
一个= 1 (NS, NS);一个= acghhmmsample (“边界条件”,NS);
转移矩阵有一个独特的平稳分布。平稳分布π是一个过渡矩阵的特征向量与特征值1。
的π= @ (x, n) ((1, n) /(眼睛(n) - x + (n))) ';
生成之前平稳分布π。
π=π(NS);
生成初始发射矩阵B
B = 0 (NS, N);为i = 1: NS B(我:)= normpdf (Y,亩(我,iloop)((我,iloop));结束
解码克隆使用一个随机的初始隐状态forward-backward算法[4]。
州(:,iloop) = acghhmmfb(π,A, B);
生成后的样品
获得每个迭代中,参数的四个街区生成如下[1]:更新块B1使用pmmh一步生成过渡矩阵,更新块B2拷贝数州使用随机抽样算法,向前传播向后更新块B3通过计算亩生成和更新块B4(开关)。
为iloop = 2: NMC%计算转换的数量从我州j为我= 1:NS为j = 1: NS st_counts总和(i, j) =((州(1:N - 1, iloop-1) = =我)。*(州(2:N, iloop-1) = = j));结束结束%更新块B1%从狄利克雷分布生成过渡矩阵C = acghhmmsample (“边界条件”st_counts + 1, NS);%计算状态概率下的平稳分布%给过渡矩阵C。照片=π(C, NS);%计算使用pmmh一步接受概率β= min ([1, exp(日志(图片(州(iloop-1)))——日志(π(州(iloop-1))))));如果兰德<β= C;π=图片;结束%更新块B2%生成拷贝数州使用向前传播,向后抽样[4]。州(:,iloop) = acghhmmfb(π,A, B);%更新块B3和B4为i = 1: NS idx_s =州(:,iloop) = =我;num_states =总和(idx_s);%如果国家我不是观测到的,然后从其先前的参数如果num_states = = 0亩(我,iloop) = acghhmmsample (“正常”mus_hyper(我),…taus_hyper(我),mu_low_bounds(我),mu_up_bounds (i));((我,iloop) = acghhmmsample (“伽马”sg_alpha(我),…sg_beta(我),sg_bounds (i));其他的Y_avg =意味着(Y (idx_s));theta_prec = 1 / taus_hyper(我)^ 2 + num_states /((我iloop-1) ^ 2;weight_means = (mus_hyper(我)/ (taus_hyper(我)^ 2)+…Y_avg * num_states /(((我iloop-1) ^ 2)) / theta_prec;%计算亩- B3亩(我,iloop) = acghhmmsample (“正常”weight_means,…1 /√(theta_prec) mu_low_bounds(我),mu_up_bounds (i));%计算(- B4Y_v =总和((Y (idx_s) -亩(我,iloop)) ^ 2);((我,iloop) = acghhmmsample (“伽马”,sg_alpha(我)+ num_states / 2,…sg_beta(我)+ Y_v / 2, sg_bounds (i));结束%更新新的亩(开关)和发射矩阵。B(我:)= normpdf (Y,亩(我,iloop)((我,iloop));结束结束
剧情后的意思μ四个州的分布。
图;为j = 1: NS次要情节(2,2,j) ksdensity(亩(j,:));标题(sprintf (“国家% d ',j))结束sgtitle (亩国家的分布);持有从;

剧情后σ四个州的分布。
图;为j = 1: NS次要情节(2,2,j) ksdensity (((j,:));标题(sprintf (“国家% d ',j))结束sgtitle (σ州的分布);持有从;

后推理
画一个状态标签为每个克隆获得的模型和计算每个状态的后验概率。
clone_states = 0 (1, N);state_prob = 0 (NS, N);state_count = 0 (NS, N);为i = 1: N%为每一个克隆状态=状态(我);为j = 1: NS state_count (j,我)=(国家= = j)总和;结束selState =找到(state_count (:, i) = = max (state_count(:,我)));如果长度(selState) > 1如果我~ = 1 clone_states (i) = clone_states(张);其他的clone_states (i) = min (selState);结束其他的clone_states (i) = selState;结束state_prob(:,我)= state_count (:, i) / NMC;结束clone_states = clone_states ';
阴谋每个克隆的状态标签样品12号染色体上PA.C.Dan.G。
图;腿= 0 (1、4);为i = 1: N如果clone_states (i) = = 1腿(1)=情节(Y (i),我“v”,“MarkerFaceColor”0.2 - 0.2 [1],…“MarkerEdgeColor”,“没有”);elseifclone_states (i) = = 2腿(2)=情节(Y (i),我“o”,“颜色”[0.4 0.4 0.4]);elseifclone_states (i) = = 3条腿(3)=情节(Y (i),我“^”,“MarkerFaceColor”(0.2 - 1 0.2),…“MarkerEdgeColor”,“没有”);elseifclone_states (i) = = 4条腿(4)=情节(Y (i),我“^”,“MarkerFaceColor”(0.2 - 0.2 (1),…“MarkerEdgeColor”,“没有”);结束持有在;结束ylims ylim (gca)传说(腿,“状态1”,“状态2”,“状态3”,“国家4”)包含(“指数”)ylabel (“Log2(比率)”)标题(“国家品牌”)举行从

全息阵列配置文件分类
对于每个密度画,生成的状态可分为局部畸变,过渡点,放大,异常值和全染色体变化[1]。在这个示例中,您会发现高层放大,过渡点和异常值12号染色体上的样本PA.C.Dan.G。
一个克隆与国家= 4被认为是高级放大[1]。找到高级放大。
high_lvl_amp_idx =找到(clone_states = = 4);
一个过渡点与得失,宣布大规模地区当改变区域的宽度超过5大型碱基对[1]。找到过渡点。
region_lim = 5 e6;focalabr_idx =[1;找到(diff (clone_states) ~ = 0); N];istranspoint = false(长度(focalabr_idx), 1);为i = 1:长度(focalabr_idx) 1 region_x = X (focalabr_idx (i + 1) - X (focalabr_idx(我));istranspoint (i + 1) = region_x > region_lim;结束trans_idx = focalabr_idx (istranspoint);%去除相邻trans_idx具有相同状态。hasadjacentstate = [diff (clone_states (trans_idx)) = = 0;真正的);trans_idx = trans_idx (~ hasadjacentstate) focalabr_idx = focalabr_idx (~ istranspoint);focalabr_idx = focalabr_idx (2: end-1);
trans_idx = 107 135 323
局外人对收益是一个焦点畸变满足它的z分数大于2,而损失一个异类的z分数小于2 [1]。
发现异常值的损失
outlier_loss_idx = focalabr_idx (clone_states (focalabr_idx) = = 1)如果~ isempty (outlier_loss_idx) [F, Xi] = ksdensity(亩(1:));[假,idx] = max (F);mu_1 = Xi (idx);[F, Xi] = ksdensity (((1:));[假,idx] = max (F);sigma_1 = Xi (idx);outlier_loss_idx = outlier_loss_idx ((Y (outlier_loss_idx)——mu_1) / sigma_1 < 2)结束
outlier_loss_idx = 0 x1空双列向量
找到异常收益
outlier_gain_idx = focalabr_idx (clone_states (focalabr_idx) = = 3);如果~ isempty (outlier_gain_idx) [F, Xi] = ksdensity(亩(3:));[假,idx] = max (F);mu_1 = Xi (idx);[F, Xi] = ksdensity (((3:));[假,idx] = max (F);sigma_1 = Xi (idx);outlier_gain_idx = outlier_gain_idx ((Y (outlier_gain_idx)——mu_1) / sigma_1 > 2)结束
outlier_gain_idx = 0 x1空双列向量
分类标签添加到样品的强度比块12号染色体PA.C.Dan.G。情节的细分意味着CBS过程进行比较。
图;hl1 =情节(X, Y,“。”,“颜色”[0.4 0.4 0.4]);持有在;如果~ isempty (high_lvl_amp_idx) hl2 =线(X (high_lvl_amp_idx), Y (high_lvl_amp_idx),…“线型”,“没有”,…“标记”,“^”,…“MarkerFaceColor”(0.2 - 0.2 (1),…“MarkerEdgeColor”,“没有”);结束如果~ isempty (trans_idx)为i = 1:元素个数(trans_idx) hl3 =线(的X (1、2) * (trans_idx(我)),[-3.5,3.5],…“线型”,“——”,…“颜色”0.6 - 0.2 [1]);结束结束如果~ isempty (outlier_gain_idx)线(X (outlier_gain_idx), Y (outlier_gain_idx),…“线型”,“没有”,…“标记”,“v”,…“MarkerFaceColor”(1 0 0),…“MarkerEdgeColor”,“没有”);结束如果~ isempty (outlier_loss_idx) hl4 =线(X (outlier_loss_idx), Y (outlier_loss_idx),…“线型”,“没有”,…“标记”,“v”,…“MarkerFaceColor”(1 0 0),…“MarkerEdgeColor”,“没有”);结束%的阴谋段意味着从哥伦比亚广播公司(CBS)过程。为i = 1:元素个数(PS.SegmentData.Start) hl5 =线([PS.SegmentData.Start (i) PS.SegmentData.End (i)),…[PS.SegmentData.Mean(我)PS.SegmentData.Mean (i)),…“颜色”(1 0 0),…“线宽”,1.5);结束ylims ylim (gca) ylabel (“Log2(比率)”)标题(sprintf (' % s -染色体% d '样本,chromID))%增加染色体12表意文字和传说故事情节。chromosomeplot (“hs_cytoBand.txt”chromID,“addtoplot”传说,gca) ([hl1 hl2, hl3, hl5],“IntensityRatio”,“放大”,…“TransitionPoint”,“CBS SegmentMean”)
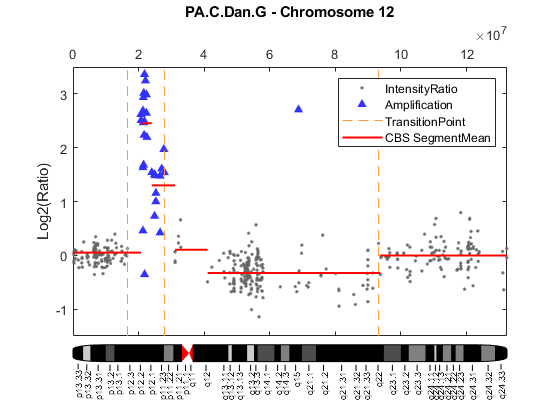
贝叶斯HMM算法发现3过渡点破碎的竖线表示的阴谋。贝叶斯HMM算法确定了两个高级放大区域,蓝色up-triangles阴谋。两个高层放大区域对应两个最小的共同区域(mcr)[2] 12号染色体上的关联与拷贝数增加解释Aguirre et al ., (2004)。贝叶斯嗯宣布第一组高强度口粮作为一个地区的高层放大。相比之下,CBS过程未能发现第二MCR和分段第一MCR分成两个区域。在本例中没有发现异常。
引用
[1],古哈。,Li, Y. and Neuberg, D., "Bayesian hidden Markov modeling of array CGH data", Journal of the American Statistical Association, 103(482):485-497, 2008.
[2]Aguirre,抗干扰,et al., "High-resolution characterization of the pancreatic adenocarcinoma genome", PNAS, 101(24):9067-72, 2004.
[3]Olshen,学士,et al., "Circular binary segmentation for the analysis of array-based DNA copy number data", Biostatistics, 5(4):557-7, 2004.
[4]沙,……,et al., "Integrating copy number polymorphisms into array CGH analysis using a robust HMM", Bioinformatics, 22(14):e431-e439, 2006