从RNA-Seq数据中识别差异表达基因
这个例子展示了如何使用负二项模型测试差异表达基因的RNA-Seq数据。
简介
RNA-Seq数据的典型差异表达分析包括归一化原始计数并进行统计检验,以拒绝或接受原假设,即两组样本在基因表达上无显著差异。这个例子展示了如何检查原始计数数据的基本统计,如何确定计数归一化的大小因素,以及如何使用负二项模型推断差异表达最大的基因。
本例的数据集包括Brooks等人描述的实验中获得的RNA-Seq数据。[1]。作者研究了siRNA敲除pasilla的影响,pasilla是一种已知的在剪接调控中发挥重要作用的基因黑腹果蝇.数据集包括对照(未处理)样本的2个生物重复和敲除(处理)样本的2个生物重复。
检查基因组特征的读计数表
RNA-Seq数据分析的起点是一个计数矩阵,其中行对应于感兴趣的基因组特征,列对应于给定的样本,值表示映射到给定样本中每个特征的读取数。
包含的文件pasilla_count_noMM.mat包含两个表,在基因水平和外显子水平的计数矩阵为每个考虑的样本。您可以使用该函数获得类似的矩阵featurecount.
负载pasilla_count_noMM.mat
预览基因的读取计数表geneCountTable (1:10,:)
ans = 10 x6表ID引用untreated3 untreated4 treated2 treated3 _______________ _________ __________ __________ ________ ________ {' FBgn0000003 '}{“3 r”}0 1 1 2{‘FBgn0000008} {2 r的}142 117 138 132{‘FBgn0000014}{“3 r”}20 12 10 19{‘FBgn0000015}{“3 r”}2 4 0 1{‘FBgn0000017} {' 3 l '} 6591 5127 4809 6027{‘FBgn0000018} {2 l的}469 530 492 574{‘FBgn0000024}{“3 r”}5 6 10 8{‘FBgn0000028} {X} 0 0 2 1{‘FBgn0000032}{“3 r”}1160 1143 1138 1415{‘FBgn0000036}{“3 r”}1 0 0 0
注意,在不进行汇总的情况下进行计数时,单个特征(在本例中为外显子)与它们的元特征分配(在本例中为基因)一起报告,然后是开始和停止位置。
%预览外显子的读取计数表exonCountTable (1:10,:)
ans = 10 x6表ID引用untreated3 untreated4 treated2 treated3 _________________________________ _________ __________ __________ ________ ________ {' FBgn0000003_2648220_2648518 '}{“3 r”}0 0 0 1{‘FBgn0000008_18024938_18025756} {2 r的}0 1 0 2{‘FBgn0000008_18050410_18051199} {2 r的}13 9 14 12{‘FBgn0000008_18052282_18052494} {2 r的}4 2 5 0{‘FBgn0000008_18056749_18058222} {2 r的}32 26日27日23日{‘FBgn0000008_18058283_18059490} {2 r的}14 18 29日22{‘FBgn0000008_18059587_18059757} {2 r的}14 3 0 {'FBgn0000008_18059821_18059938'} {'2R'} 0 0 2 0 {'FBgn0000015_12758093_12760298'} {'3R'} 1 2 0 0 {'FBgn0000017_16615461_16618374'} {'3L'} 1807 1572 1557 1702
您可以通过创建一个如下所示的逻辑向量来注释和分组示例:
samples = geneCountTable(:,3:end).Properties.VariableNames;未经处理= strncmp(样本,“未经处理”长度(“未经处理”))处理= strncmp(样品,“治疗”长度(“治疗”))
未处理= 1x4逻辑阵列1 1 0 0处理= 1x4逻辑阵列0 0 1 1
绘制特征分配图
所包含的文件还包含一个表geneSummaryTable使用已分配和未分配SAM条目的摘要。通过考虑分配给给定基因组特征(例如外显子或基因)的读取数,以及未分配的读取数(即不重叠任何特征)或不明确的读取数(即重叠多个特征),可以绘制出计数结果的基本分布。
st = geneSummaryTable({“分配”,“Unassigned_ambiguous”,“Unassigned_noFeature”}:)栏(table2array (st)”,“堆叠”);传奇(st.Properties.RowNames ',“翻译”,“没有”,“位置”,“东南”);包含(“样本”) ylabel (“读取次数”)
st = 3x4表untreated3 untreated4 treated2 treated3 __________ __________ __________ __________ Assigned 1.5457e+07 1.6302e+07 1.5146e+07 1.8856e+07 unassignd_ambiguous 1.5708e+05 1.6882e+05 1.6194e+05 1.9977e+05 unassignd_nofeature 7.5455e+05 5.8309e+05 5.8756e+05 6.8356e+05

注意,SAM文件中的一小部分对齐记录没有在汇总表中报告。您可以从SAM文件中的记录总数与同一SAM文件在计数过程中处理的记录总数之间的差异中注意到这一点。这些未报告的记录对应于映射到引用序列的记录,这些引用序列在GTF文件中没有注释,因此在计数过程中没有处理。如果基因模型包含读映射步骤中使用的所有参考序列,那么所有记录将在汇总表的一个类别中报告。
geneSummaryTable {“TotalEntries”:} - sum(geneSummaryTable{2:end,:})
Ans = 89516 95885 98207 104629
绘图读取覆盖给定的染色体
在不使用函数进行汇总的情况下执行读计数featurecount,默认id由属性或元特征(默认为gene_id)和特征的开始和停止位置(默认为外显子)组成。您可以使用外显子起始位置来绘制任何考虑到的染色体的读覆盖,例如染色体臂2L。
考虑染色体臂2Lchr2L = strcmp(exonCountTable.)参考,' 2 l ');exonCount = exonCountTable{:,3:end};%检索外显子起始位置exonStart = regexp(exonCountTable{chr2L,1},“_ (\ d +) _”,“令牌”);exonStart = [exonStart{:}];exonStart = cellfun(@str2num, [exonStart{:}]');%按起始位置排序外显子[~,idx] = sort(exonStart);沿着基因组坐标的% plot读取覆盖率图;情节(exonStart (idx) exonCount (idx、处理),“。r”,...exonStart (idx) exonCount (idx,未经处理的),“。”);包含(“基因组的位置”);ylabel (“读取计数(外显子级别)”);标题(“在染色体臂2L上读取计数”);为每一组分别绘制阅读覆盖率图图;次要情节(2,1,1);情节(exonStart (idx) exonCount (idx,未经处理的),“。r”);ylabel (“读取计数(外显子级别)”);标题(染色体臂2L(处理过的样本));次要情节(2,1,2);情节(exonStart (idx) exonCount (idx、处理),“。”);ylabel (“读取计数(外显子级别)”);包含(“基因组的位置”);标题(“染色体臂2L(未处理样本)”);


或者,您也可以根据给定染色体中每个基因的起始位置绘制读取覆盖图。该文件pasilla_geneLength.mat包含一个表,其中包含相应基因注释文件中每个基因的起始和停止位置。
%负载基因启动和停止位置信息负载pasilla_geneLengthgeneLength (1:10,:)
ans = 10 x5表ID名称引用起停 _______________ ___________ _________ _____ _____ {' FBgn0037213}{‘CG12581} 3 r 380 10200{‘FBgn0000500}{“Dsk”}3 r 15388 16170{‘FBgn0053294}{‘CR33294} 3 r 17136 21871{‘FBgn0037215}{‘CG12582} 3 r 23029 30295{‘FBgn0037217}{‘CG14636} 3 r 30207 41033{‘FBgn0037218}{“辅助”}3 r 37505 53244{‘FBgn0051516}{‘CG31516} 3 r 44179 45852{‘FBgn0261436}{‘DhpD} 3 r 53106 54971{‘FBgn0037220}{‘CG14641} 3 r 56475 58077{‘FBgn0015331}{“abs”}3 r58765 60763
考虑3号染色体(“参考”是一个分类变量)chr3 = (geneLength。)参考= =' 3 l '| (genlength .)参考= =“3 r”);总和(chr3)考虑3号染色体上的基因数量。counts = geneCountTable{:,3:end};[~,j,k] = intersect(geneCountTable{:,“ID”}, geneLength {chr3,“ID”});gstart = geneLength{k,“开始”};Gcounts = counts(j,:);%按升序开始位置排序[~,idx] = sort(gstart);%图读取覆盖基因组位置图;情节(gstart (idx) gcounts (idx、处理),“。r”,...gstart (idx) gcounts (idx,未经处理的),“。”);包含(“基因组的位置”) ylabel (“读计数”);标题(“读取3号染色体的计数”);
Ans = 6360

规范化读计数
RNA-Seq数据中的读取计数已被发现与转录本[2]的丰度呈线性相关。然而,给定基因的读取数不仅取决于基因的表达水平,还取决于测序的读取总数和基因转录本的长度。因此,为了从读取计数中推断一个基因的表达水平,我们需要考虑测序深度和基因转录本长度。规范化读计数的一种常见技术是使用RPKM (read Per Kilobase Mapped)值,其中读计数由生成的读总数(以百万为单位)和每个转录本的长度(以千基为单位)规范化。然而,这种归一化技术并不总是有效的,因为很少的,非常高表达的基因可以支配整个lane计数和扭曲表达分析。
一种更好的归一化技术是通过考虑每个样本的大小因子来计算有效库大小。通过将每个样本的计数除以相应的大小因子,我们将所有计数值放在一个共同的尺度上,使它们具有可比性。直观地说,如果样本A的测序深度是样本B的N倍,那么即使在表达上没有差异,样本A的无差异表达基因的读计数预计也会比样本B的平均高N倍。
为了估计大小因素,取观察计数与伪参考样本计数比值的中位数,其计数可以通过考虑所有样本[3]中每个基因的几何平均值来获得。然后,为了将观察到的计数转换为一个公共尺度,将每个样本中的观察到的计数除以相应的大小因子。
%估计伪参考与几何平均逐行pseudoRefSample =几何学(计数,2);nz = pseudoRefSample > 0;ratio = bsxfun(@rdivide,counts(nz,:),pseudoRefSample(nz));sizeFactors = median(ratio,1)
sizeFactors = 0.9374 0.9725 0.9388 1.1789
%转化为共同尺度normCounts = bsxfun(@rdivide,counts,sizeFactors);normCounts (1:10,:)
Ans = 1.0e+03 * 0 0.0010 0.0011 0.0017 0.1515 0.1203 0.1470 0.1120 0.0213 0.0123 0.0107 0.0161 0.0021 0.0041 0 0.0008 7.0315 5.2721 5.1225 5.1124 0.5003 0.5450 0.5241 0.4869 0.0053 0.0062 0.0107 0.0068 00 0.0021 0.0008 1.2375 1.1753 1.2122 1.2003 000 0.0008
您可以通过使用函数来欣赏这种归一化的效果箱线图表示统计指标,如中位数、四分位数、最小值和最大值。
图;次要情节(2,1,1)maboxplot (log2(计数)“标题”,“原始读取计数”,“定位”,“水平”) ylabel (“样本”)包含(“log2(计数)”maboxplot(log2(normCounts),“标题”,“规范化读计数”,“定位”,“水平”) ylabel (“样本”)包含(“log2(计数)”)

计算均值、色散和折叠变化
为了更好地表征数据,我们考虑归一化计数的均值和离散度。读取计数的方差由两个项的和给出:跨样本的方差(原始方差)和通过计数读取(炮噪声或泊松)测量表达式的不确定度。原始方差项在高表达基因中占主导地位,而在低表达基因中则占主导地位。您可以将经验离散值与归一化计数的平均值绘制成对数刻度,如下所示。
考虑平均数=均值(normCounts(:,已处理),2);=平均值(normCounts(:,未处理),2);%考虑分散性dishandled = std(normCounts(:, handled),0,2) ./dispunhandled = std(normCounts(:,未处理),0,2)./%的对数-对数刻度图图;重对数(meanTreated dispTreated,“r”。);持有在;重对数(meanUntreated dispUntreated,“b”。);包含(“log2(的意思)”);ylabel (“log2(分散)”);传奇(“治疗”,“未经处理”,“位置”,“西南”);

由于重复的数量很少,因此预期散布值在真实值周围有一些方差是不足为奇的。其中一些方差反映了采样方差,而另一些则反映了样本中基因表达的真实变异性。
你可以通过计算每个基因的fold change (FC),即处理组的计数与未处理组的计数之比,来观察两种条件下基因表达的差异。一般来说,这些比值都是用log2的尺度来考虑的,因此任何变化都是与零对称的(例如,1/2或2/1的比值在对数尺度中对应的是-1或+1)。
%计算平均值和log2FCmeanBase = (meanprocessed + meanprocessed) / 2;foldChange = meanhandled ./ mean未处理;log2FC = log2(foldChange);平均百分比图与折叠变化(MA图)mairplot (meanTreated meanUntreated,“类型”,“马”,“Plotonly”,真正的);集(get (gca),“包含”),“字符串”,“归一化计数的平均数”甘氨胆酸)组(get (,“Ylabel”),“字符串”,“log2(褶皱变化)”)
警告:忽略零值

方法可以用相应的基因名称注释图中的值,交互式地选择基因,并将基因列表导出到工作空间mairplot函数如下所示:
mairplot (meanTreated meanUntreated,“标签”, geneCountTable。ID,“类型”,“马”);
警告:忽略零值

将每个基因的均值和折叠变化信息存储在表格中,方便快捷。然后,通过按基因名称为表建立索引,您可以访问关于一个给定基因或一组满足特定标准的基因的信息。
%创建关于每个基因的统计数据表geneTable = table(meanBase, meanprocessed, mean未处理,foldChange,log2FC);geneTable.Properties.RowNames = genecountable . id;
%总结总结(geneTable)
变量:meanBase: 11609x1 double值:Min 0.21206中位数201.24 Max 2.6789e+05 meantreatment: 11609x1 double值:Min 0中位数201.54 Max 2.5676e+05 mean processed: 11609x1 double值:Min 0中位数199.44 Max 2.7903e+05 foldChange: 11609x1 double值:Min 0中位数0.99903 Max Inf log2FC: 11609x1 double值:Min -Inf中位数-0.001406 Max Inf
%预览geneTable (1:10,:)
ans = 10x5表meanBase meanTreated mean未处理foldChange log2FC ________ ___________ _____________ __________ ___________ FBgn0000003 0.9475 1.3808 0.51415 2.6857 1.4253 FBgn0000008 132.69 129.48 135.9 0.95277 -0.069799 FBgn0000014 15.111 13.384 16.838 0.79488 -0.33119 FBgn0000014 15.111 13.384 16.838 0.79488 -0.33119 FBgn0000015 1.7738 0.42413 3.1234 0.13579 -2.8806 FBgn0000017 5634.6 5117.4 6151.8 0.83186 -0.26559 FBgn0000018 514.08 505.48 522.67 0.96711 -0.048243 FBgn0000024 7.2354 8.7189 5.752 1.5158 0.60009 FBgn0000028 0.74465 1.48930 Inf Inf FBgn0000032 1206.3 1206.2 1206.4 0.99983 -0.00025093 FBgn0000036 0.21206 0.42413 0 Inf Inf
访问特定基因的信息myGene =“FBgn0261570”;geneTable (myGene:) geneTable (myGene, {“meanBase”,“log2FC”})%访问关于给定基因列表的信息myGeneSet = {“FBgn0261570”,“FBgn0261573”,“FBgn0261575”,“FBgn0261560”};geneTable (myGeneSet:)
ans = 1x5 table meanBase meanprocessed mean未处理foldChange log2FC ________ ___________ _____________ __________ _______ FBgn0261570 4435.5 4939.1 3931.8 1.2562 0.32907 ans = 1x2 table meanBase log2FC ________ _______ FBgn0261570 4435.5 0.32907 ans = 4x5 table meanBase meanprocessed mean未处理foldChange log2FC ________ ___________ _____________ __________ _______ FBgn0261570 4435.5 4939.1 3931.8 1.2562 0.32907 FBgn0261573 2936.9 2954.8 2919.1 1.0122 0.01753 FBgn0261575 4.3776 5.6318 3.12341.8031 0.85047 FBgn0261560 2041.1 1494.3 2588 0.57738 -0.7924
用负二项模型推导微分表达式
确定两种条件下的基因表达是否在统计上不同,包括拒绝原假设,即两个数据样本来自具有相等均值的分布。该分析假设读取计数是根据负二项分布建模的(如[3]中提出的)。这个函数nbintest用三种可能的选项执行这种假设检验,以指定方差和均值之间的联系类型。
通过指定方差和均值之间的联系作为恒等式,我们假设方差等于均值,计数由泊松分布[4]建模。
id = nbintest(counts(:,已处理),counts(:,未处理),“VarianceLink”,“身份”);h = plotVarianceLink(tIdentity);%设置自定义标题h (1) .Title。字符串=“处理样本的方差链接”;h (2) .Title。字符串=“未处理样本的方差链接”;


或者,通过将方差指定为抛丸噪声项(即平均值)和常数乘以平均值的平方之和,计数将根据[5]中描述的分布建模。常数项使用数据中的所有行进行估计。
tConstant = nbintest(counts(:,已处理),counts(:,未处理),“VarianceLink”,“不变”);h = plotVarianceLink(tConstant);%设置自定义标题h (1) .Title。字符串=“处理样本的方差链接”;h (2) .Title。字符串=“未处理样本的方差链接”;


最后,将方差考虑为炮点噪声项(即均值)和均值的局部回归非参数平滑函数之和,根据[3]中提出的分布对计数进行建模。
tLocal = nbintest(counts(:,已处理),counts(:,未处理),“VarianceLink”,“LocalRegression”);h = plotVarianceLink(tLocal);%设置自定义标题h (1) .Title。字符串=“处理样本的方差链接”;h (2) .Title。字符串=“未处理样本的方差链接”;


为了评估哪个适合考虑的数据是最好的,您可以在单个图中比较拟合曲线,如下所示。
h = plotVarianceLink(tLocal,“比较”,真正的);%设置自定义标题h (1) .Title。字符串=“处理样本的方差链接”;h (2) .Title。字符串=“未处理样本的方差链接”;


的输出nbintest包含一个p值的向量。p值表示在零假设下,基因表达发生与观察到的(甚至更强)一样强的变化的概率,即条件对基因表达没有影响。在p值的直方图中,我们观察到低值的富集(由于差异表达基因),而其他值则均匀分布(由于无差异表达基因)。值等于1的富集是由于计数非常低的基因造成的。
图;直方图(tLocal.pValue 100)包含(“假定值”) ylabel (“频率”)标题(“假定值浓缩”)

过滤掉那些计数相对较低的基因,观察在(0,1)范围内非显著p值的更均匀分布。注意,这并不影响显著p值的分布。
lowCountThreshold = 10;lowCountGenes = all(count < lowCountThreshold, 2);直方图(tLocal.pValue (~ lowCountGenes), 100)包含(“假定值”) ylabel (“频率”)标题(“无低计数基因的p值富集”)

多次测试和调整p值
由于多重测试问题,用阈值p值来确定哪些折叠变化比其他变化更显著并不适用于这类数据分析。在执行大量同时进行的测试时,仅由于偶然的原因而获得显著结果的概率会随着测试数量的增加而增加。为了考虑多重测试,对p值进行修正(或调整),使至少一个由于偶然而观察到的显著结果的概率保持在期望的显著性水平以下。
Benjamini-Hochberg (BH)调整[6]是一种统计方法,它提供一个调整后的p值,回答以下问题:如果调整后的p值低于给定阈值的所有基因都被认为是显著的,那么假阳性的比例将是多少?将调整后的p值设置为0.1的阈值,相当于认为10%的假阳性是可以接受的,并通过考虑调整后的p值低于该阈值的所有基因来确定显著表达的基因。
%计算调整后的p值(BH校正)padj = mafdr(tLocal.pValue,“BHFDR”,真正的);%添加到现有表geneTable。pvalue = tLocal.pValue;geneTable。Padj = Padj;创建一个有重要基因的表格sig = geneTable。Padj < 0.1;geneTableSig = geneTable(sig,:);geneTableSig = sortrows(基因表lesig,“padj”);numberSigGenes = size(genetableig,1)
numberSigGenes = 1904
识别上调和下调最多的基因
现在,你可以通过考虑所选截止点以上的绝对倍数变化来确定上调或下调最多的基因。例如,log2尺度中1的截止值产生了2倍变化上调的基因列表。
%发现上调基因up = geneTableSig。log2FC > 1;upGenes = sortrows(geneTableSig(up,:),“log2FC”,“下”);numberSigGenesUp = sum(up)%显示前10个上调基因top10GenesUp = upGenes(1:10,:)找到被下调的基因down = geneTableSig。log2FC < -1;downGenes = sortrows(geneTableSig(down,:),“log2FC”,“提升”);numberSigGenesDown = sum(down)%发现前10个下调基因top10GenesDown = downGenes(1:10,:)
numberSigGenesUp = 129 top10GenesUp = 10x7 table meanBase meanTreated mean未处理foldChange log2FC pvalue padj ________ ___________ _____________ __________ ______ __________ __________ FBgn0030173 3.3979 6.7957 0 Inf Inf 0.0063115 0.047764 FBgn0036822 3.1364 6.2729 0 Inf Inf 0.012203 0.079274 FBgn0052548 8.158 15.269 1.0476 14.575 3.8654 0.00016945 0.0022662 FBgn0050495 6.8315 12.635 1.0283 12.287 3.6191 0.0018949 0.017972 FBgn0063667 20.573 38.042 3.1042 12.255 3.6153 8.5037e-08 2.3845e-06FBgn0033764 91.969 167.61 16.324 10.268 3.3601 1.8345e-25 2.9174e-23 FBgn0037290 85.845 155.46 16.228 9.5801 3.26 3.5583e-23 4.6941e-21 FBgn0033733 7.4634 13.384 1.5424 8.6773 3.1172 0.0027276 0.024283 FBgn0037191 7.1766 12.753 1.6003 7.9694 2.9945 0.0047803 0.038193 FBgn0033943 6.95 12.319 1.581 7.7921 2.962 0.0053635 0.041986 numberSigGenesDown = 181 top10GenesDown = 10x7 table meanBase meanTreated mean未处理foldChange log2FC pvalue padj ________ ___________ _____________ _________________ __________ __________ FBgn0053498 15.469 0 30.938 0 -Inf 9.8404e-11 4.345e-09 FBgn0259236 6.618 0 -Inf 1.5526e-05 0.00027393 fbgn0039383 4.3703 0 8.7405 0 -Inf 0.00066783 0.0075343 FBgn0039331 3.6954 0 7.3908 0 -Inf 0.0019558 0.018474 FBgn0040697 3.419 0 6.8381 0 -Inf 0.0027378 0.024336 FBgn0034972 2.9145 0 5.8291 0 -Inf 0.0068564 0.05073 FBgn0040967 2.6382 0 5.2764 0 -Inf 0.0096039 0.065972 FBgn0031923 2.3715 0 4.7429 0 -Inf 0.016164 0.098762 FBgn0085359 62.473 2.9786 121.970.024421 -5.3557 5.5813e-33 1.5068e-30 FBgn0004854 7.4674 0.53259 14.402 0.03698 -4.7571 8.1587e-05 0.0012034
通过在对数尺度上绘制折叠变化与均值的关系,并根据调整后的p值对数据点着色,可以很好地显示基因表达及其意义。
图散射(log2 (geneTable.meanBase) geneTable.log2FC 3, geneTable.padj,“o”) colormap(flipud(cool(256))) colorbar;ylabel (“log2(褶皱变化)”)包含('log2(归一化计数的平均值)')标题(“罗斯福的改变”)
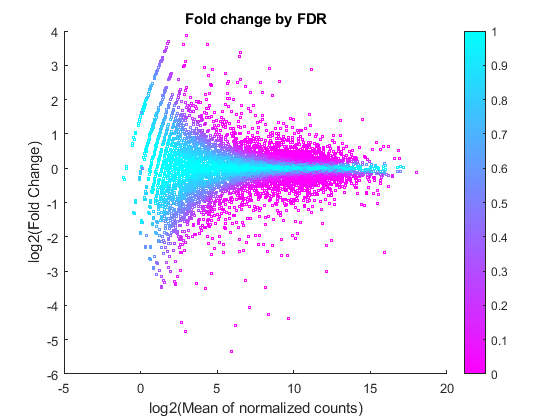
你可以在这里看到,对于弱表达的基因(即那些低均值的基因),FDR通常是高的,因为低读计数是由泊松噪声主导的,因此任何生物可变性都淹没在读计数的不确定性中。
参考文献
布鲁克斯等。果蝇和哺乳动物RNA调控图谱的保存。基因组研究2011。21:193 - 202。
Mortazavi等。利用RNA-Seq定位和量化哺乳动物转录组。自然方法2008。5:621 - 628。
安德斯等。序列计数数据的微分表达式分析。基因组生物学2010。11: R106。
Marioni等。RNA-Seq:技术重现性评估和与基因表达阵列的比较。基因组研究2008。18:1509 - 1517。
罗宾逊等。评估标签丰度差异的调节性统计检验。2007年生物信息学。23日(21):2881 - 2887。
[6]本杰明等。控制错误发现率:多重测试的一种实用而强大的方法。1995.皇家统计学会学报,B57系列(1):289-300。
另请参阅
featurecount|mairplot|nbintest|plotVarianceLink
相关的话题
您也可以从以下列表中选择网站: