基因表达谱分析
这个例子显示了一个数量的基因表达谱中寻找模式的方法。
探索数据集
这个示例使用的数据发布的微阵列基因表达研究酵母DeRisi, et al . 1997 [1]。作者使用DNA微阵列研究颞几乎所有基因的基因表达酿酒酵母在呼吸的代谢转变,从发酵。表达水平测量时间点7点在diauxic转变。完整的数据集可以从基因表达综合网站,下载https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28。
的MAT-fileyeastdata.mat包含表达式的值(log2比率CH2DN_MEAN和CH1DN_MEAN)从七个步骤在这个实验中,基因的名称,一个数组时代的表达水平的测定。
负载yeastdata.mat
获得一个想法的大小可以使用的数据元素个数(基因)显示有多少基因包含在数据集。
元素个数(基因)
ans = 6400
您可以访问索引与实验相关的基因名称的变量基因细胞数组代表基因的名字。例如,15元素基因YAL054C。这表明的第15行变量yeastvalues包含YAL054C的表达水平。
基因{15}
ans = ' YAL054C '
一个简单的情节可以用来表示这个开放的表达谱。
情节(次yeastvalues(15日:))包含(的时间(小时));ylabel (Log2相对表达水平的);

你也可以画出实际表达比率,而不是log2-transformed值。
情节(*,2。^ yeastvalues(15日:))包含(的时间(小时));ylabel (的相对表达水平的);

与这相关的基因ORF, ACS1,似乎强烈上调在diauxic转变。你可以比较这个基因的表达其他基因的表达通过绘制多个行是基于相同的数据。
持有在情节(*,2。^ yeastvalues(: 16:26)”)包含(的时间(小时));ylabel (的相对表达水平的);标题(概要文件表达水平的);

过滤的基因
通常,基因表达数据集包括信息对应的基因,在实验中不显示任何有趣的变化。使它更容易找到有趣的基因,可以减少数据集的大小一个子集,只包含最重要的基因。
如果你浏览基因列表,你会看到几个点标记为“空”。这些是空点阵列,而他们可能会与他们相关的数据,对于本示例中,您可以考虑这些点噪声。这些点可以使用比较字符串函数和从数据集中删除索引的命令。
emptySpots = strcmp (“空”,基因);yeastvalues (emptySpots:) = [];基因(emptySpots) = [];元素个数(基因)
ans = 6314
在数据集也有看到几个地方被标记为表达水平南。这表明没有数据收集这个地方在特定的时间步。处理这些缺失值的一种方法是归咎于他们使用数据均值或中位数为特定基因随着时间的推移。这个示例使用一个不那么严格的方法简单地扔掉任何基因的数据在一个或多个表达水平没有测量。这个函数isnan是用来识别基因缺失的数据和索引命令用来消除基因缺失的数据。
nanIndices =任何(isnan (yeastvalues), 2);yeastvalues (nanIndices:) = [];基因(nanIndices) = [];元素个数(基因)
ans = 6276
如果你情节的表达谱剩下的配置文件,你会看到,大多数配置文件是平的,而不是明显不同于其他人。这个平面数据显然是使用的,因为它表明,与这些概要文件相关的基因并不影响diauxic转变;然而,在这个例子中,您感兴趣的基因表达发生巨大变化,陪diauxic转变。您可以使用过滤功能的生物信息学工具箱™删除基因与各种类型的配置文件不提供有用的信息基因受到代谢变化的影响。
您可以使用genevarfilter函数过滤掉基因差异小。函数返回一个逻辑数组(即。相同大小的面具)的变量基因相应的行yeastvalues与方差大于0和第十百分位对应于那些低于阈值。你可以用面具来索引过滤和删除的值的基因。
掩码= genevarfilter (yeastvalues);:yeastvalues = yeastvalues(面具);基因=基因(面具);元素个数(基因)
ans = 5648
这个函数genelowvalfilter删除基因有非常低的绝对表达式值。注意,这些过滤功能还可以自动计算过滤数据和名字,所以没有必要指标原始数据使用的面具。
(面具,yeastvalues,基因)= genelowvalfilter (yeastvalues,基因,“absval”log2 (3));元素个数(基因)
ans = 822
最后,您可以使用该功能geneentropyfilter删除基因的概要低熵,例如熵水平在15百分位的数据。
(面具,yeastvalues,基因)= geneentropyfilter (yeastvalues,基因,“prctile”15);元素个数(基因)
ans = 614
聚类分析
现在,您已经有了一个可控的基因列表,您可以使用一些寻找配置文件之间的关系不同的集群技术统计和机器学习的工具箱™。层次聚类的功能pdist计算两两之间的距离的概要文件和链接创建一个层次聚类树。
corrDist = pdist (yeastvalues,“相关系数”);clusterTree =连杆(corrDist,“平均”);
的集群函数计算集群基于截止距离或最大数量的集群。在这种情况下,maxclust选项是用来确定16个不同的集群。
集群=集群(clusterTree“maxclust”16);
基因在这些集群的配置文件可以使用一个简单的循环和绘制在一起次要情节命令。
图为c = 1:16次要情节(4 4 c);情节(次,yeastvalues ((: = = c)集群)');轴紧结束sgtitle (概要文件的层次聚类);
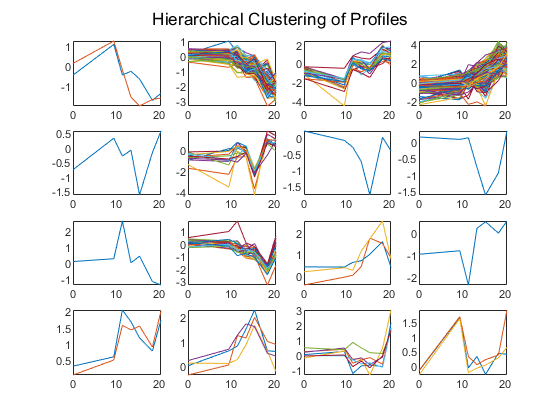
统计和机器学习工具箱也有k - means聚类函数。十六个集群被发现,但由于算法不同的这些不一定是相同的集群层次聚类发现的。
初始化随机数发生器的状态,以确保这些命令生成的数据匹配数据的HTML版本,这个例子。
rng (“默认”);[cidx,点击率数据]= kmeans (yeastvalues 16“距离”,“相关系数”,“代表”5,“disp”,“最后一次”);图为c = 1:16次要情节(4 4 c);情节(次,yeastvalues ((cidx = = c):) ');轴紧结束sgtitle (“k - means聚类的配置文件”);
复制1,21个迭代,总额= 23.4699的距离。复制2,22个迭代,总额= 23.5615的距离。复制3,10次迭代,总额= 24.823的距离。复制4,28个迭代,总额= 23.4501的距离。复制5,19个迭代,总额= 23.5109的距离。最好的总金额= 23.4501的距离

而不是策划所有的概要,您可以只质心的阴谋。
图为c = 1:16次要情节(4 4 c);情节(次,点击率数据(c:) ');轴紧轴从结束sgtitle (“k - means聚类的配置文件”);

您可以使用clustergram函数创建一个热图的表达水平和系统树图的输出层次聚类。
cgObj = clustergram (yeastvalues(:, 2:结束),“RowLabels”的基因,“ColumnLabels”,乘以(2:结束));

主成分分析
主成分分析(PCA)是一个有用的技术,可以用来降低大型数据集的维数,比如从微阵列。主成分分析也可以用来在嘈杂的数据找到信号。这个函数mapcaplot计算一个数据集的主成分和创建散点图的结果。可以交互式地选择数据点的情节,和这些点会自动突出显示在另一个阴谋。这允许您可视化同时多个维度。
h = mapcaplot (yeastvalues,基因);

注意,散点图的前两个主成分得分表明,有两种截然不同的区域。这不是意外的过滤过程去除很多方差较低或低的基因信息。这些基因会出现在散点图的中间。
如果你想看的主成分值,主成分分析统计和机器学习的工具箱函数是用来计算一个数据集的主成分。
[pc, zscores pcvars] = pca (yeastvalues);
第一个输出,个人电脑是一个矩阵的主成分yeastvalues数据。矩阵的第一列是第一主成分,第二列是第二主成分,等等。第二个输出,zscores,由主成分得分。,a representation of yeastvalues in the principal component space. The third output,pcvars,包含主成分方差,这给多少的衡量数据的方差所占的每个主要组件。
很明显,第一主成分占多数的方差模型。可以计算的精确比例的方差占每个组件,如下所示。
pcvars. /笔(pcvars) * 100
ans = 79.8316 9.5858 4.0781 2.6486 2.1723 0.9747 - 0.7089
这意味着近90%的方差占了前两个主成分。您可以使用cumsum命令查看累计方差之和。
cumsum (pcvars. /笔(pcvars) * 100)
ans = 79.8316 89.4174 93.4955 96.1441 98.3164 99.2911 - 100.0000
如果你想要更多的控制策划的主要组件,您可以使用散射函数。
图散射(zscores (: 1), zscores (:, 2));包含(“第一主成分”);ylabel (第二主成分的);标题(的主成分散点图);

另一种方法来创建一个散点图是与功能gscatter从统计和机器学习工具。gscatter创建一个分组每组散点图,分有不同的颜色和标志。您可以使用clusterdata,或任何其他集群功能,对点进行分组。
图pcclusters = clusterdata (zscores (:, 1:2),“maxclust”8“链接”,“影音”);gscatter (zscores (: 1) zscores (:, 2), pcclusters, hsv(8)包含(“第一主成分”);ylabel (第二主成分的);标题(“主成分散点图与彩色集群”);

自组织映射
如果你有深度学习工具箱™,你可以使用一个自组织映射(SOM)集群数据。
%检查深度学习工具箱安装如果~((存在“selforgmap”),“文件”)disp (这个例子的自组织映射部分需要深度学习工具箱。)返回结束
的selforgmap函数创建一个新的SOM网络对象。这个例子将生成一个SOM使用前两个主成分。
P = zscores (: 1:2)”;网= selforgmap ([4 4]);
列车网络使用缺省参数。
网=火车(净,P);
使用plotsom显示网络数据的散点图。注意,SOM算法使用随机起始点从运行结果会有所不同。
图绘制(P (1:)、P (2:)“.g”,“markersize”,20)在net.layers plotsom (net.iw {1}, {1} .distances)从

您可以指定集群使用SOM找到最近的节点数据集内的每个点。
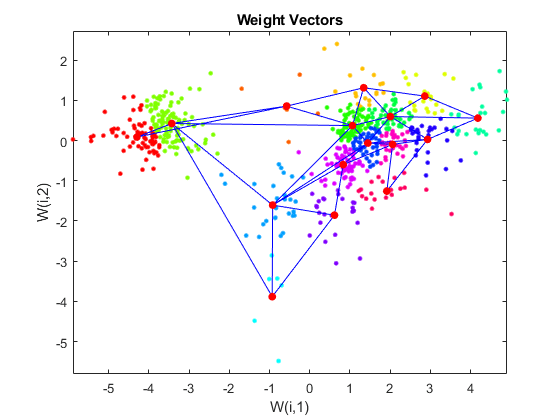
距离= dist (P’, net.IW {1}”);[d, cndx] = min(距离,[],2);% cndx包含集群指数图gscatter (P (1:)、P (2:), cndx, hsv(元素个数(独特(cndx))));传说从;持有在net.layers plotsom (net.iw {1}, {1} .distances);持有从
关闭所有的数据。
关闭(“所有”);删除(cgObj);删除(h);
引用
[1]DeRisi J.L.,Iyer, V.R. and Brown, P.O., "Exploring the metabolic and genetic control of gene expression on a genomic scale", Science, 278(5338):680-6, 1997.