主要内容
序列分析
基因组和蛋白质组学序列,对准和系统源
通过对核苷酸或氨基酸序列进行分析来增强对序列特征,功能和演化的更深理解。使用成对或多个序列对齐方法比较序列。计算序列属性和统计数据,以获得更多关于数据的物理,化学和生物学特性的洞察力。对在线或本地数据库中的已知序列进行爆炸搜索。通过从序列的成对距离构建系统发育树来确定生物体之间的进化关系。
- 数据导入和导出
从公共存储库和本地文件系统导入序列数据,包括Fasta,Genbank,Genpept,Embl,Blast,PDB,PFAM,Clustalw,GCG,Phylip,Newick和FastQ;写入各种格式,包括Fasta,PDB和Newick - 核苷酸序列分析
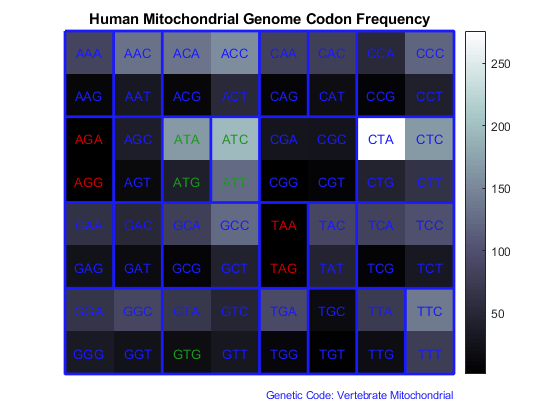
计算和交互式探索序列统计;计算序列属性;分析主题;设计底漆;找到限制酶 - 蛋白质和氨基酸序列分析
计算和交互式探索氨基酸序列统计;计算序列属性;寻找切割酶 - 序列对齐
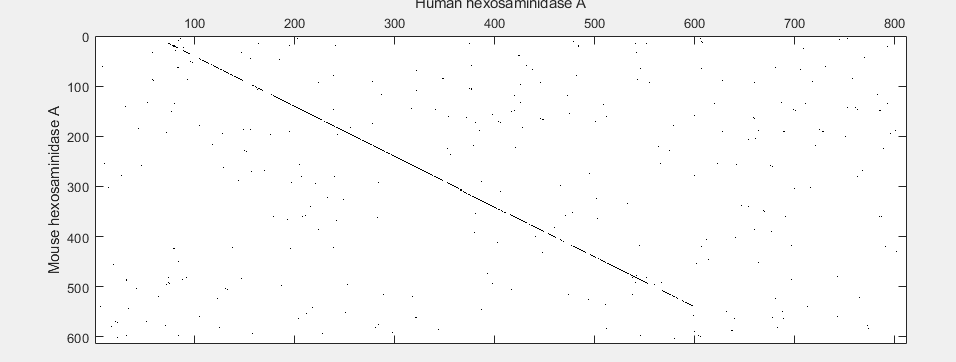
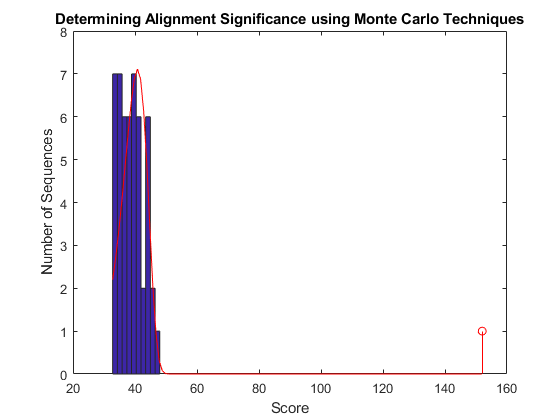
使用动态编程算法多个,成对和简档序列对齐;爆炸搜索和对齐;标准和定制评分矩阵 - 系统发育分析
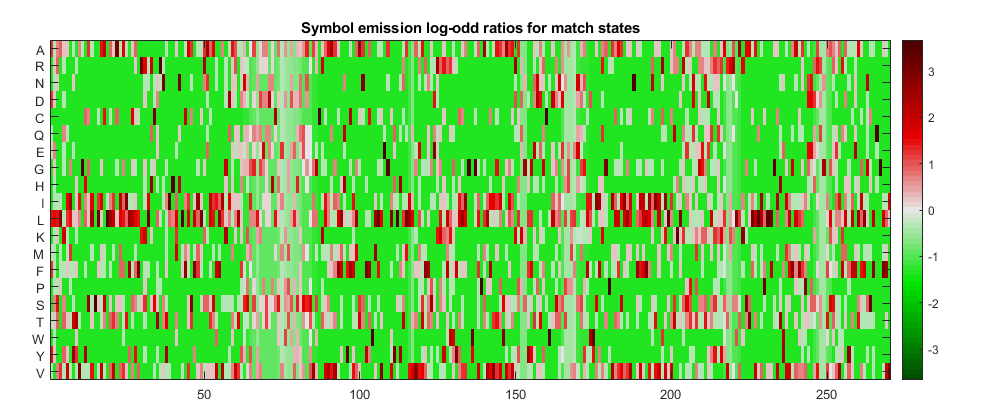
重建,观看,互动和编辑系统发育树;自动启动方法置信评估;同义词分析
特色例子
您还可以从以下列表中选择一个网站: